Bioequivalence
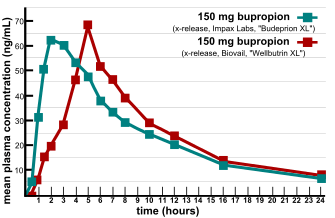
Bioequivalence is a term in pharmacokinetics used to assess the expected in vivo biological equivalence of two proprietary preparations of a drug. If two products are said to be bioequivalent it means that they would be expected to be, for all intents and purposes, the same.
Birkett (2003) defined bioequivalence by stating that, "two pharmaceutical products are bioequivalent if they are pharmaceutically equivalent and their bioavailabilities (rate and extent of availability) after administration in the same molar dose are similar to such a degree that their effects, with respect to both efficacy and safety, can be expected to be essentially the same. Pharmaceutical equivalence implies the same amount of the same active substance(s), in the same dosage form, for the same route of administration and meeting the same or comparable standards."[1]
The United States Food and Drug Administration (FDA) has defined bioequivalence as, "the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study."[2]
Bioequivalence
In determining bioequivalence, for example, between two products such as a commercially available Brand product and a potential to-be-marketed Generic product, pharmacokinetic studies are conducted whereby each of the preparations are administered in a cross-over study to volunteer subjects, generally healthy individuals but occasionally in patients. Serum/plasma samples are obtained at regular intervals and assayed for parent drug (or occasionally metabolite) concentration. Occasionally, blood concentration levels are neither feasible or possible to compare the two products (e.g. inhaled corticosteroids), then pharmacodynamic endpoints rather than pharmacokinetic endpoints (see below) are used for comparison. For a pharmacokinetic comparison, the plasma concentration data are used to assess key pharmacokinetic parameters such as area under the curve (AUC), peak concentration (Cmax), time to peak concentration (Tmax), and absorption lag time (tlag). Testing should be conducted at several different doses, especially when the drug displays non-linear pharmacokinetics.
In addition to data from bioequivalence studies, other data may need to be submitted to meet regulatory requirements for bioequivalence. Such evidence may include:
- analytical method validation
- in vitro-in vivo correlation studies(IVIVC)
Regulatory definition
Australia
In Australia, the Therapeutics Goods Administration (TGA) considers preparations to be bioequivalent if the 90% confidence intervals (90% CI) of the rate ratios, between the two preparations, of Cmax and AUC lie in the range 0.80-1.25. Tmax should also be similar between the products.[1]
There are tighter requirements for drugs with a narrow therapeutic index and/or saturable metabolism – thus no generic products exist on the Australian market for digoxin or phenytoin for instance.
Europe
According to regulations applicable in the European Economic Area[3] two medicinal products are bioequivalent if they are pharmaceutically equivalent or pharmaceutical alternatives and if their bioavailabilities after administration in the same molar dose are similar to such a degree that their effects, with respect to both efficacy and safety, will be essentially the same. This is considered demonstrated if the 90% confidence intervals (90% CI) of the ratios for AUC0-t and Cmax between the two preparations lie in the range 80.00 – 125.00%.
United States
The FDA considers two products bioequivalent if the 90% CI of the relative mean Cmax, AUC(0-t) and AUC(0-∞) of the test (e.g. generic formulation) to reference (e.g. innovator brand formulation) should be within 80.00% to 125.00% in the fasting state. Although there are a few exceptions, generally a bioequivalent comparison of Test to Reference formulations also requires administration after an appropriate meal at a specified time before taking the drug, a so-called "fed" or "food-effect" study. A food-effect study requires the same statistical evaluation as the fasting study, described above.[2]
See also
References
- 1 2 Birkett DJ (2003). "Generics - equal or not?" (PDF). Aust Prescr. 26: 85–7.
- 1 2 Center for Drug Evaluation and Research (2003). "Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products — General Considerations" (PDF). United States Food and Drug Administration.
- ↑ Committee for Medicinal Products for Human Use (20 January 2010). "Guideline on the Investigation of Bioequivalence" (pdf). European Medicines Agency. Retrieved 21 April 2011.
Further reading
- Hussain AS, et al. The Biopharmaceutics Classification System: Highlights of the FDA's Draft Guidance Office of Pharmaceutical Science, Center for Drug Evaluation and Research, Food and Drug Administration.
- Mills D (2005). Regulatory Agencies Do Not Require Clinical Trials To Be Expensive International Biopharmaceutical Association: IBPA Publications.
- FDA CDER Office of Generic Drugs – further U.S. information on bioequivalence testing and generic drugs