COSMO-RS
COSMO-RS (short for COnductor like Screening MOdel for Real Solvents)[1] is a quantum chemistry based equilibrium thermodynamics method with the purpose of predicting chemical potentials µ in liquids. It processes the screening charge density σ on the surface of molecules to calculate the chemical potential µ of each species in solution. As an initial step a quantum chemical COSMO[2] calculation for all molecules is performed and the results (e.g. the screening charge density) are stored in a database. In a separate step COSMO-RS uses the stored COSMO results to calculate the chemical potential of the molecules in a liquid solvent or mixture. The resulting chemical potentials are the basis for other thermodynamic equilibrium properties such as activity coefficients, solubility, partition coefficients, vapor pressure and free energy of solvation. The method was developed to provide a general prediction method with no need for system specific adjustment.
Due to the use of σ from COSMO calculations, COSMO-RS does not require functional group parameters. Quantum chemical effects like group-group interactions, mesomeric effects and inductive effects also are incorporated into COSMO-RS by this approach.
The COSMO-RS method was first published in 1995 by A. Klamt.[1] A refined version of COSMO-RS was published in 1998 [3] and is the basis for newer developments and reimplementations.[4][5][6][7][8]
Basic principles
The below description is a simplified overview of the COSMO-RS version published in 1998.
Assumptions
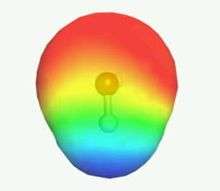
- The liquid state is incompressible
- All parts of the molecular surfaces can be in contact with each other
- Only pairwise interactions of molecular surface patches are allowed
As long as the above assumptions hold, the chemical potential µ in solution can be calculated from the interaction energies of pairwise surface contacts.
COSMO-RS equations
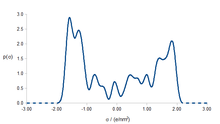
Within the basic formulation of COSMO-RS, interaction terms depend on the screening charge density σ. Each molecule and mixture can be represented by the histogram p(σ), the so-called σ-profile. The σ-profile of a mixture is the weighted sum of the profiles of all its components. Using the interaction energy Eint(σ,σ') and the σ-profile of the solvent p(σ'), the chemical potential µs(σ) of a surface piece with screening charge σ is determined as:
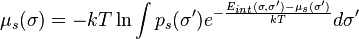
Due to the fact that µs(σ) is present on both sides of the equation, it needs to be solved iteratively. By combining the above equation with px(σ) for a solute x, and adding the σ-independent combinatorial and dispersive contributions, the chemical potential for a solute X in a solvent S results in:
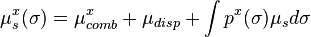
In analogy to activity coefficient models used in chemical engineering, such as NRTL, UNIQUAC or UNIFAC, the final chemical potential can be split into a combinatorial and a residual (non ideal) contribution. The interaction energies Eint(σ,σ') of two surface pieces are the crucial part for the final performance of the method and different formulations are used within the various implementations. In addition to the liquid phase terms a chemical potential estimate for the ideal gas phase µgas has been added to COSMO-RS to enable the prediction of vapor pressure, free energy of solvation and related quantities.
Interaction energy (Residual)
The residual part is the sum of three different contributions, where Emisfit and Ehb are part of Eint and µdisp is added directly to the chemical potential.
Electrostatic interaction
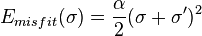
In the Emisfit expression α is an adjustable parameter and σ and σ' refer to the screening charge densities of the two surface patches in contact. This term has been labeled "misfit" energy, because it results from the mismatch of the charged surface pieces in contact. It represents the Coulomb interaction relative to the state in a perfect conductor. A molecule in a perfect conductor (COSMO state) is perfectly shielded electronically; each charge on the molecular surface is shielded by a charge of the same size but of opposite sign. If the conductor is replaced by surface pieces of contacting molecules the screening of the surface will not be perfect any more. Hence an interaction energy from this misfit of σ on the surface patches will arise.
Hydrogen bonding energy
![E_{hb} (\sigma)=c_{hb}(T)\max[0,\sigma_{acc}-\sigma_{hb}] \min[0,\sigma_{don}+\sigma_{hb}]](../I/m/eee9eb8ef4134615b1f939ce1b3a16d4.png)
In the Ehb expression σacc and σdon are the screening charge densities of the hydrogen bond acceptor and donor respectively. The hydrogen bonding threshold σhb and the prefactor chb are adjustable parameters. The max[] and min[] construction ensures that the screening charge densities of the acceptor and donor exceeds the threshold for hydrogen bonding.
Dispersion (van der Waals energy)
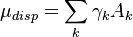
The COSMO-RS dispersion energy of a solute depends on an element (k) specific prefactor γ and the amount of exposed surface A of this element. It is not part of the interaction energy but enters the chemical potential directly.
Parameters
Though the use of quantum chemistry reduces the need for adjustable parameters, some fitting to experimental data is inevitable. The basic parameters are α, chb, σhb as used in the interaction energies, and one general parameter for the effective contact area. In addition, one adjustable van der Waals parameter γ per element is required. All parameters either are general or element specific, which is a distinctive feature of COSMO-RS as compared to group contribution methods like UNIFAC.
See also
References
- 1 2 "Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena", A. Klamt, J. Phys. Chem., 99, 2224-2235 (1995), DOI: 10.1021/j100007a062
- ↑ "COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and its Gradient", A. Klamt and G. Schüürmann, J. Chem. Soc. Perkin Trans. II 799-805 (1993) DOI: 10.1039/P29930000799
- ↑ "Refinement and Parametrization of COSMO-RS", A. Klamt, V. Jonas, T. Bürger and J. C. W. Lohrenz, J. Phys. Chem. A 102, 5074-5085 (1998), DOI: 10.1021/jp980017s
- ↑ "A Priori Phase Equilibrium Prediction from a Segment Contribution Solvation Model", S.-T. Lin and S.I. Sandler, Ind. Eng. Chem. Res., 41 (5), 899–913 (2002), DOI: 10.1021/ie001047w
- ↑ "Performance of a Conductor-Like Screening Model for Real Solvents Model in Comparison to Classical Group Contribution Methods", H. Grensemann and J. Gmehling, Ind. Eng. Chem. Res., 44 (5), 1610–1624 (2005), DOI:10.1021/ie049139z
- ↑ "Infinite Dilution Activity Coefficients for Trihexyltetradecyl Phosphonium Ionic Liquids: Measurements and COSMO-RS Prediction", T. Banerjee and A. Khanna , J. Chem. Eng. Data, 51 (6), 2170–2177 (2006), DOI:10.1021/je0602925
- ↑ "An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package. Part II. COSMO for real solvents", C.C. Pye, T. Ziegler, E. van Lenthe, J.N. Louwen, Can. J. Chem. 87, 790 (2009), DOI: 10.1139/V09-008
- ↑ "On the influence of basis sets and quantum chemical methods on the prediction accuracy of COSMO-RS", R. Franke, B. Hannebauer, Phys. Chem. Chem. Phys., 13, 21344-21350 (2011), DOI: 10.1039/C1CP22317H
Overviews / Reviews
"COSMO-RS: From quantum Chemistry to Fluid Phase Thermodynamics and Drug Design", A. Klamt, Elsevier: Amsterdam, 2005, ISBN 978-0444519948
"COSMO-RS: An Alternative to Simulation for Calculating Thermodynamic Properties of Liquid Mixtures", A. Klamt, F. Eckert and W. Arlt, Annual Review of Chemical and Biomolecular Engineering, 1, 101-122, (2010), DOI: 10.1146/annurev-chembioeng-073009-100903