Living anionic polymerization
Living anionic polymerization is a living polymerization technique involving an anionic propagating species.
Living anionic polymerization was demonstrated by Szwarc and co workers in 1956. Their initial work was based on the polymerization of styrene and dienes. One of the remarkable features of living anionic polymerization is that the mechanism involves no formal termination step. In the absence of impurities, the carbanion would still be active and capable of adding another monomer. The chains will remain active indefinitely unless there is inadvertent or deliberate termination or chain transfer. This gave rise to two important consequences:
- The number average molecular weight, Mn, of the polymer resulting from such a system could be calculated by the amount of consumed monomer and the initiator used for the polymerization, as the degree of polymerization would be the ratio of the moles of the monomer consumed to the moles of the initiator added.
-
![M_n = M_o \frac {[\mbox{M}]_o} {[\mbox{I}]}](../I/m/e89c6c9353ba02d0828240aedd9376a9.png) , where Mo = formula weight of the repeating unit, [M]o = initial concentration of the monomer, and [I] = concentration of the initiator.
, where Mo = formula weight of the repeating unit, [M]o = initial concentration of the monomer, and [I] = concentration of the initiator.
-
- All the chains are initiated at roughly the same time. The final result is that the polymer synthesis can be done in a much more controlled manner in terms of the molecular weight and molecular weight distribution (Poisson distribution).
The following experimental criteria have been proposed as a tool for identifying a system as living polymerization system.
- Polymerization until the monomer is completely consumed and until further monomer is added.
- Constant number of active centers or propagating species.
- Poisson distribution of molecular weight
- Chain end functionalization can be carried out quantitatively.
However, in practice, even in the absence of terminating agents, the concentration of the living anions will reduce with time due to a decay mechanism termed as spontaneous termination.[1]
Synthesis of complex architectures
Polymerization reactions excluding a termination and transfer step are particularly useful for the synthesis of functionalized polymers with well defined architectures.[2] The consumption of the monomer results in stable, anionic polymer chain ends, allowing reactions with a variety of electrophilic functional groups post polymerization.

However the efficacy of these reactions depends on a number of variables such as chain-end structure, solvent, temperature, and concentration. Alternatively, by controlling the functionality of the initiator, one can prepare polymers having different geometries such as symmetric and asymmetric stars, comb shaped, etc.
Block copolymers
Synthesis of block copolymers by sequential monomer addition is one of the most important applications of living polymerization as it offers the best control over structure. The nucleophilicity of the resulting carbanion will govern the order of monomer addition, as the monomer forming the less nucleophilic propagating species may inhibit the addition of the more nucleophilic monomer onto the chain. An extension of the above concept is the formation of triblock copolymers where each step of such a sequence aims to prepare a block segment with predictable, known molecular weight and narrow molecular weight distribution without chain termination or transfer.[3]

Sequential monomer addition is the dominant method in the literature, mainly due to its simplicity. Nevertheless, also this concept has serious limitations. For example, for a given set of three monomers (A, B, and C), sequential living anionic polymerization can afford all possible linear BC structures (i.e., ABC, ACB, and BAC)exclusively in the highly unlikely scenario that the following requirements are fulfilled: (a) similar monomer reactivity, (b) high crossover efficiency, and (c) absence of side reactions.
Chemoselective stepwise coupling between living polymers and a heterobifunctional linking agent is a versatile, facile, and efficient methodology for block copolymer synthesis. This protocol is based on the quantitative formation of the end-functionalized precursor in the first coupling step. The applicability range is becoming exceptionally wide upon transformation reactions, e.g., by tailoring the chain-termini reactivity via endcapping. This novel protocol considerably expands the limits of sequential monomer addition, rendering viable the construction of advanced and tailor-made materials via an effortless pathway.(http://www3.interscience.wiley.com/journal/114280481/abstract) Moreover, this strategy, enables synthesis of linear block copolymer structures that are not accessible via sequential monomer addition. Even for common A-b-B structures, sequential block copolymerization gives access to well defined block copolymers only if the crossover reaction rate constant is significantly higher than the rate constant of the homopolymerization of the second monomer, i.e., kAA >> kBB. http://www3.interscience.wiley.com/journal/121628088/abstract
Star Shaped Polymers

A star shaped polymer is a polymeric structure in which several chains emanate from a single junction point known as the core. The control offered by anionic polymerization makes it a very popular pathway to synthesize molecules with such complex geometry. It allows quantitative studies of the degree of branching of the polymer on the overall properties of the substance.
One of the first pathways explored was the use of a multifunctional initiator, but it was limited by the insolubility of such compounds and there was no control over the reactivity of each branch. A second, more efficient way was proposed - the addition of a multifunctional electrophilic terminator at the end of the polymerization of a linear polymer. This is analogous to the convergent synthesis of dendrimers, and is efficient as long as the stoichiometry between the terminating agent and the starting monomers are maintained.
The third route is through the addition of small amounts of cross – linking agents to the polymeric precursor (for example, addition of divinyl benzene to polystyryl lithium). There are three prime reactions that take place:
- The crossover of the polystyryl chain to divinylbenzene
- The block copolymerization of divinylbenzene
- The reaction of the pendant vinyl groups of divinyl benzene with linear polystyryl branches
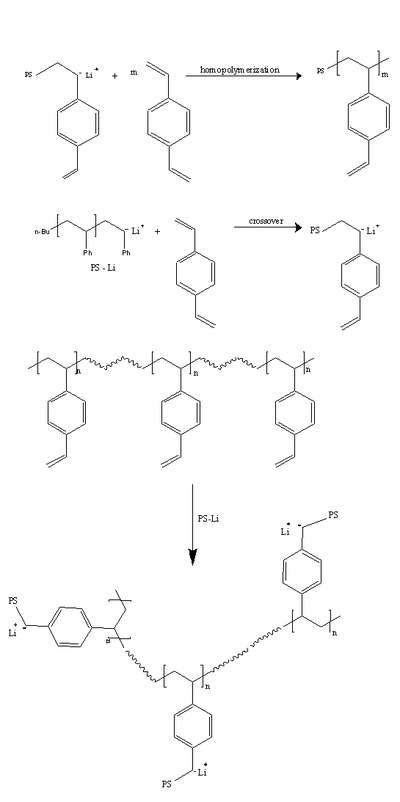
The uniformity in the structure is a function of the rate of the crossover reaction compared to the other two reactions. The number of branches of the star molecules cannot be precisely predicted, as it is a complex function of the reaction variables. For example, the amount of divinyl benzene added in the above pathway, compared to the number of active chains, is a key factor governing the overall degree of branching of the polymer.
The method is widely used for the synthesis of star shaped polystyrene with divinyl benzene with low molecular weight distributions.[4] Star shaped polymethyl methacrylate was similarly synthesized using ethylene glycol dimetthacrylate as a crosslinker.[5] The molecular weights obtained are comparatively high (~40kDa), which is thought to be necessary to avoid the gelation due to inter core reactions. The protection of the core groups by the branches gave such polymers the name "porcupine polymers".[6]
End-Group Functionalization
Living anionic polymerization can also be used to incorporate functional groups. These end-groups are usually added post-polymerization. End-groups that have been used in the functionalization of α-haloalkanes include hydroxide, -NH2, -OH, -SH, -CHO,-COCH3, -COOH, and epoxides.
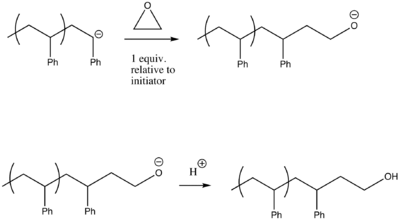
An alternative approach for functionalizing end-groups is to begin polymerization with a functional anionic initiator.[7] In this case, the functional groups are protected since the ends of the anionic polymer chain is a strong base. This method leads to polymers with controlled molecular weights and narrow molecular weight distributions.[8]
References
- ↑ Odian, G. Ionic Chain Polymerization;In Principles of Polymerization; Wiley-Interscience: Staten Island, New York, 2004, pp. 372-463.
- ↑ Cowie, J.; Arrighi,V. Polymers: Chemistry and Physics of Modern Materials; CRC Press: Boca Raton, FL, 2008.
- ↑ Hsieh, H.;Quirk, R. Anionic Polymerization: Principles and practical applications; Marcel Dekker, Inc: New York, 1996.
- ↑ Hadjichristidis, N.; Iatrou, H.; Pitsikalis, P.; Mays, J. Macromolecular architectures by living and controlled/living polymerizations. Prog. Polym. Sci. 2006, 31, pp.1068-1132. DOI =10.1016/j.progpolymsci.2006.07.002
- ↑ Efstratiadis, V.; Tselikas, Y.; Hadjichristidis, N.; Li, J.; Yunan, W.; Mays, J. Synthesis and characterization of poly(methyl methacrylate) star polymers Polym Int.1994, 4, pp. 171-179. doi=10.1002/pi.1994.210330208
- ↑ Rempp, P.; Franta, E.; Herz, J. Macromolecular Engineering by Anionic Methods. Adv. Polym. Sci. 1998, 4, pp. 145-173 DOI=10.1007/BFb0025273
- ↑ Hong K.; Uhrig, D.;Mays, J. Living Anionic Polymerization. Curr Opin Solid State Mater Sci. 1999,4, 531-538. doi=10.1016/S1359-0286(00)00011-5
- ↑ Quirk, R. Anionic Polymerization. In Encyclopedia of Polymer Science and Technology; John Wiley and Sons: New York, 2003.