Somatic hypermutation
Somatic hypermutation (or SHM) is a cellular mechanism by which the immune system adapts to the new foreign elements that confront it (e.g. microbes), as seen during class switching. A major component of the process of affinity maturation, SHM diversifies B cell receptors used to recognize foreign elements (antigens) and allows the immune system to adapt its response to new threats during the lifetime of an organism.[1] Somatic hypermutation involves a programmed process of mutation affecting the variable regions of immunoglobulin genes. Unlike germline mutation, SHM affects only an organism's individual immune cells, and the mutations are not transmitted to the organism's offspring.[2]
Mistargeted somatic hypermutation is a likely mechanism in the development of B-cell lymphomas.[3]
Targeting
When a B cell recognizes an antigen, it is stimulated to divide (or proliferate). During proliferation, the B cell receptor locus undergoes an extremely high rate of somatic mutation that is at least 105-106 fold greater than the normal rate of mutation across the genome.[2] Variation is mainly in the form of single base substitutions, with insertions and deletions being less common. These mutations occur mostly at “hotspots” in the DNA, which are concentrated in hypervariable regions. These regions correspond to the complementarity determining regions; the sites involved in antigen recognition on the immunoglobulin.[4] The "hotspots" of somatic hypermutation vary depending on the base that is being mutated. RGYW for a G, WRCY for a C, WA for an A and TW for a T.[5][6] The overall result of the hypermutation process is achieved by a balance between error-prone and high fidelity repair.[7] This directed hypermutation allows for the selection of B cells that express immunoglobulin receptors possessing an enhanced ability to recognize and bind a specific foreign antigen.[1]
Mechanisms

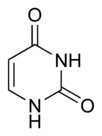
Experimental evidence supports the view that the mechanism of SHM involves deamination of cytosine to uracil in DNA by an enzyme called Activation-Induced (Cytidine) Deaminase, or AID.[8][9] A cytosine:guanine pair is thus directly mutated to a uracil:guanine mismatch. Uracil residues are not normally found in DNA, therefore, to maintain the integrity of the genome, most of these mutations must be repaired by high-fidelity Base excision repair enzymes. The uracil bases are removed by the repair enzyme, uracil-DNA glycosylase.[9] Error-prone DNA polymerases are then recruited to fill in the gap and create mutations.[8][10]
The synthesis of this new DNA involves error-prone DNA polymerases, which often introduce mutations at the position of the deaminated cytosine itself or neighboring base pairs. During B cell division the immunoglobulin variable region DNA is transcribed and translated. The introduction of mutations in the rapidly proliferating population of B cells ultimately culminates in the production of thousands of B cells, possessing slightly different receptors and varying specificity for the antigen, from which the B cell with highest affinities for the antigen can be selected. The B cells with the greatest affinity will then be selected to differentiate into plasma cells producing antibody and long-lived memory B cells contributing to enhanced immune responses upon reinfection.[2]
The hypermutation process also utilizes cells that auto-select against the 'signature' of an organism's own cells. It is hypothesized that failures of this auto-selection process may also lead to the development of an auto-immune response.
See also
References
- 1 2 Janeway, C.A., Travers, P., Walport, M., Shlomchik, M.J. (2005). Immunobiology (6th ed.). Garland Science. ISBN 0-8153-4101-6.
- 1 2 3 Oprea, M. (1999) Antibody Repertoires and Pathogen Recognition: The Role of Germline Diversity and Somatic Hypermutation (Thesis) University of Leeds.
- ↑ Odegard V.H.; Schatz D.G. (2006). "Targeting of somatic hypermutation". Nat. Rev. Immunol. 6 (8): 573–583. doi:10.1038/nri1896. PMID 16868548.
- ↑ Li, Z., Wool, C.J., Iglesias-Ussel, M.D., Ronai, D., and Scharff, M.D. (2004). "The generation of antibody diversity through somatic hypermutation and class switch recombination". Genes & Development. 18 (1): 1–11. doi:10.1101/gad.1161904. PMID 14724175.
- ↑ Dunn-Walters, DK; Dogan, A; Boursier, L; MacDonald, CM; Spencer, J (1998). "Base-specific sequences that bias somatic hypermutation deduced by analysis of out of frame genes.". J. Immunol. 160: 2360–64.
- ↑ Spencer, J; Dunn-Walters, DK (2005). "Hypermutation at A-T base pairs: The A nucleotide replacement spectrum is affected by adjacent nucleotides and there is no reverse complimentarity of sequences around A and T nucleotides.". J. Immunol. 175: 5170–77. doi:10.4049/jimmunol.175.8.5170.
- ↑ Liu, M., Schatz, D.G. (2009). "Balancing AID and DNA repair during somatic hypermutation. Trends in Immunology". Trends in Immunology. 30 (4): 173–181. doi:10.1016/j.it.2009.01.007. PMID 19303358.
- 1 2 Teng, G.; Papavasiliou, F.N. (2007). "Immunoglobulin Somatic Hypermutation". Annu. Rev. Genet. 41: 107–120. doi:10.1146/annurev.genet.41.110306.130340. PMID 17576170.
- 1 2 Larson, E.D.; Maizels, N. (2004). "Transcription-coupled mutagenesis by the DNA deaminase AID". Genome Biol. 5 (3): 211. doi:10.1186/gb-2004-5-3-211. PMC 395756
 . PMID 15003109.
. PMID 15003109. - ↑ Bachl, J., Ertongur, I., Jungnickel, B. (2006). "Involvement of Rad18 in somatic hypermutation". Proc. Natl. Acad. Sci. USA. 103 (32): 12081–86. doi:10.1073/pnas.0605146103.
External links
- Immunoglobulin somatic hypermutation at the US National Library of Medicine Medical Subject Headings (MeSH)