Vinylcyclopropane rearrangement
The vinylcyclopropane rearrangement or vinylcyclopropane-cyclopentene rearrangement is a ring expansion reaction, converting a vinyl-substituted cyclopropane ring into a cyclopentene ring.[1][2][3]

Intense experimental as well as computational investigations have revealed that mechanistically, the vinylcyclopropane rearrangement can be thought of as either a diradical-mediated two-step and/or orbital-symmetry-controlled pericyclic process. The amount by which each of the two mechanisms is operative is highly dependent on the substrate.
Due to its ability to form cyclopentene rings the vinylcyclopropane rearrangement has served several times as a key reaction in complex natural product synthesis.
Origins and history
In 1959, a young research chemist with Humble Oil and Refining (Esso, now Exxon) named Norman P. Neureiter was instructed to find new uses for the excess butadiene produced from one of the refinery processes. Discussions about carbene chemistry with one of the company's most respectable consultants at the time, William von Eggers Doering, then a professor at Yale, led the young Ph.D. graduate from Northwestern University to follow a recent procedure combining both, carbenes and butadiene.[4] In particular the procedure described the reaction of 1,3-butadiene with carbenes generated from the action of base on chloroform or bromoform, which had been studied previously by Doering.[5] Neureiter then took the resulting 1,1-dichloro-2,2-dimethylcyclopropane and under pyrolysis conditions (above 400 °C) discovered a rearrangement to 4,4-dichlorocyclopentene which today is considered to be the first thermal vinylcyclopropane-cyclopentene rearrangement in history.[6]
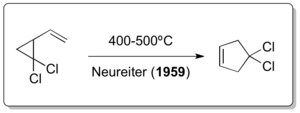
The corresponding all-carbon version of the reaction was independently reported by Emanuel Vogel[7] and Overberger & Borchert just one year after the Neureiter publication appeared.[8][9] Interestingly Doering, although actively interacting with Humble Oil and Refining - and therefore also with Neureiter - as a consultant, in a 1963 publication stated the following : "CREDIT for discovery that vinylcyclopropane rearranges to cyclopentene is due to Overberger and Borchert, and Vogel et al., who appear to have developed several examples of the rearrangement independently."[10] The development of further vinylcyclopropane rearrangement variants didn't take long as demonstrated by Atkinson & Rees in 1967,[11] Lwowski in 1968.[12] and Paladini & Chuche in 1971.[13]
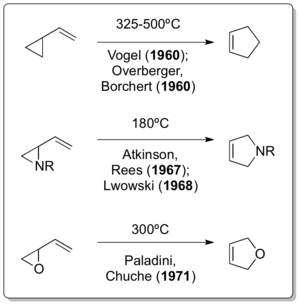
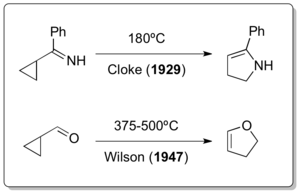
It is remarkable that the classical vinylcyclopropane rearrangement was discovered after two of its heteroatom variants had already been reported for almost 30 years and 12 years, respectively. Although it is believed that the vinylcylcopropane rearrangement must have occurred during Nikolay Demyanov's preparation of vinylcyclopropane by Hofmann elimination at elevated temperatures in 1922[14] the cyclopropylimine-pyrroline rearrangement by Cloke in 1929[15] and Wilson's cyclopropylcarbaldehyde-2,3-dihydrofuran rearrangement in 1947[16] are really the only examples of vinylcyclopropane-like rearrangements.
Mechanism
The mechanistic discussion on whether the vinylcyclopropane rearrangement proceeds through a diradical-mediated two-step or a fully concerted orbital-symmetry-controlled mechanism has been going on for more than half a century. Kinetic data together with the secondary kinetic isotope effects observed at the vinyl terminus of the vinylcyclopropane suggest a concerted mechanism whereas product distribution indicates a stepwise-diradical mechanism.[17] In the 1960s, shortly after the rearrangement was discovered, it was established that the activation energy for the vinylcyclopropane rearrangement is around 50 kcal/mol.[18] The kinetic data obtained for this rearrangement were consistent with a concerted mechanism where cleavage of the cyclopropyl carbon-carbon bond was rate-limiting. Albeit a concerted mechanism seemed likely it was shortly recognized that the activation energy to break the carbon-carbon bond in unsubstituted cyclopropane was with 63 kcal/mol[19] exactly 13 kcal/mol higher in energy than the parent activation energy, a difference remarkably similar to the resonance energy of the allyl radical.[20] Immediately people started to appreciate the possibility for a diradical intermediate arising from homolytic cleavage of the weak C1-C2-cyclopropane bond under thermal conditions.
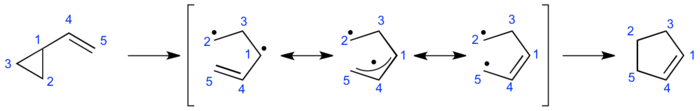
The discussion on whether the vinylcyclopropane rearrangement proceeds via a fully concerted or a two-step, non-concerted mechanism was given further careful consideration when Woodward and Hoffmann used the vinylcyclopropane rearrangement to exemplify [1,3]-sigmatropic concerted alkyl shifts in 1969.[21] They hypothesized that if a concerted mechanism was operative the consequences of orbital-symmetry controlled factors would only allow the formation of certain products. According to their analysis of a vinylcyclopropane substituted with three R groups the antarafacial [1,3]-shift of bond 1,2 to C-5, with retention at C-2, leading to the ar cyclopentene and the suprafacial [1,3]-shift of bond 1,2 to C-5, with inversion at C-2, leading to cyclopentene si are symmetry allowed whereas the suprafacial [1,3]-shift of bond 1,2 to C-5, with retention at C-2, leading to cyclopentene sr and the antarafacial [1,3]-shift of bond 1,2 to C-5, with inversion at C-2, leading to the ai cyclopentene are symmetry-forbidden. It is important to note that Woodward and Hoffmann based their analysis solely on the principles of the conservation of orbital symmetry theory without however making any mechanistic or stereochemical prediction.
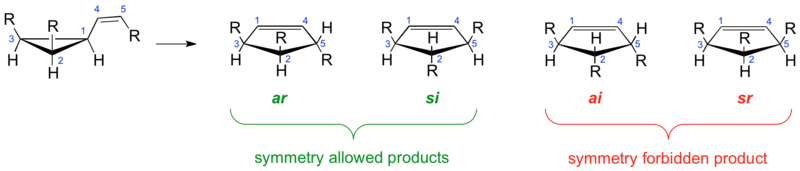
The attention directed towards the vinylcyclopropane rearrangement by Woodward and Hoffmann as a representative example for [1,3]-carbon shifts clearly enhanced the interest in this reaction. Furthermore, their analysis revealed potential experiments that would allow to distinguish between a concerted or stepwise mechanism. The stereochemical consequences of a concerted reaction pathway on the reaction outcome suggested an experiment where one would correlate the obtained reaction stereochemistry with the predicted reaction stereochemistry for a model substrate. Observing the formation of ai- and sr-cyclopentene products would support the notion that a stepwise, non-concerted mechanism is operative whereas their absence would point towards a fully concerted mechanism. As it turned out finding an appropriate substituted model substrate to study the stereochemical outcome of the vinylcyclopropane rearrangement was much more challenging than initially thought since side reaction such as the homodienyl [[[1,5]-hydrogen shift]]s and more so thermal stereomutations tend to scramble stereochemical distinctions much faster than rearrangements lead to the cyclopentene products.

Even though deconvolution of the complex kinetic scenarios underlying these rearrangements was difficult there have been several studies reported where exact and explicit deconvolutions of kinetic and stereochemical raw data to account for the stereochemical contributions arising from competitive stereomutations was possible.[22][23][24][25][26][27][28][29][30][31][31]
Thereby rate constants for all four stereochemically distinct pathways of the vinylcyclopropane rearrangement could be determined.
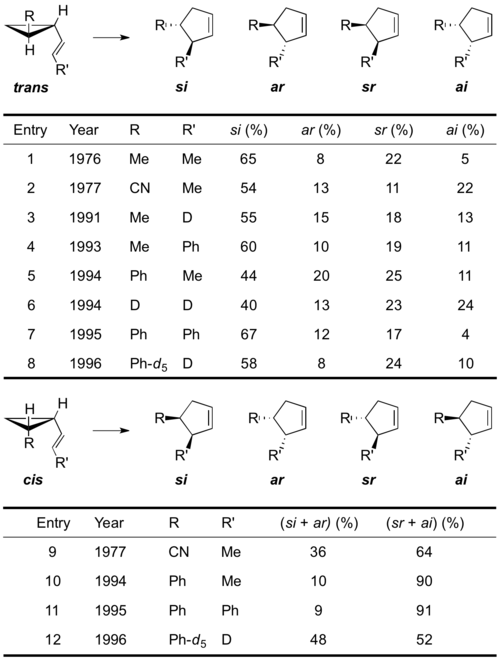
The data clearly indicated that the mechanistic preferences of the rearrangements are system dependent. Whereas trans-vinylcyclopropanes tend to form more of the symmetry-allowed ar- and si-cyclopentenes supportive of a concerted mechanism, the cis-vinylcyclopropanes preferentially yield the symmetry-forbidden ai- and sr- products suggesting a more stepwise, diradical mechanism. The influence of substituent effects on the reaction stereochemistry also becomes apparent from the data. Substituents with increased radical stabilizing ability not only lower the rearrangements activation energy but also reclosure of the initially formed diradical species becomes slower relative to the rate of cyclopentene formation resulting in an overall more concerted mechanism with less stereomutation (e.g. entry 6 & 7). In all cases though all the four products were formed indicating that both orbital-symmetry controlled pericyclic, as well as diradical-mediated two-step mechanisms are operative either way. The data is consistent with the formation of biradical species on a relatively flat potential energy surface allowing for restricted conformational flexibility before the products are formed. The amount of conformational flexibility and therefore conformational evolution accessible to the diradical species before forming product depends on the constitution of the potential energy surface. This notion is also supported by computational work.[32] One transition state with a high diradicaloid character was found. Following the potential energy surface of the lowest energy path of the reaction it was found that a very shallow regime allows the diradical species to undergo conformational changes and stereoisomerization reactions with minor energetic consequences. Furthermore, it was shown that substituents can favor stereoselective pathways by destabilizing species that allow stereochemical scrambling.
Methodology development
Arguably the biggest drawback of the vinylcyclopropane rearrangement as a synthetic method is its intrinsically high activation barrier resulting in very high reaction temperatures (500-600 °C). Not only do these high temperatures allow side reactions with similar activation energies, such as homodienyl-[[[1,5]-hydrogen shifts]], to occur but also do they significantly limit the functional groups tolerated in the substrates. It was well recognized by the chemical community that in order for this reaction to become a useful synthetic method, hopefully applicable in complex natural product settings at some point, some reaction development had to be done. Some of the earliest attempts to improve the vinylcyclopropane rearrangement as a synthetic method came from the Corey group in 1972.[33] They found that the reaction temperature could be lowered drastically when the cyclopropane ring contained a dithiane group. Even though the dithiane-substituted vinylcyclopropane substrates required two synthetic steps starting from the corresponding 1,3-dienes the method proved itself successful for the synthesis of a variety of substituted cyclopentenes. The immediate rearrangement products could be easily converted to the corresponding cyclopentenones.

Only a year later Simpson and co-workers demonstrated that also simple methoxy-substituted vinylcyclopropanes show significantly faster reaction rates allowing the rearrangement to take place at 220 °C.[34]

A big improvement came in the mid-1970s from Barry M. Trost's group. It was found that siloxyvinylcyclopropanes[35] as well as the analogous sulfinylvinylcyclopropanes[36] could be used as substrates to build interesting annulated cyclopentene structures. Albeit these reactions still required reaction temperatures above 300 °C they were able to make really useful products arising from the annulation of cyclopentene to a present ring system.

Paquette demonstrated that vinylcyclopropane rearrangements can also be mediated photochemically.[37][38] In a particularly intriguing example he was able to show that vinylcyclopropanes embedded within a cyclooctane core can be converted to the corresponding [5-5]-fused ring systems.

Further reaction improvement came when Hudlicky[39] and Brown[40] proved that vinylcyclopropane rearrangements are amenable to transition metal catalysts. Using a Rh(I) acetate catalyst they were able to promote rearrangements from room temperature to 80 °C.
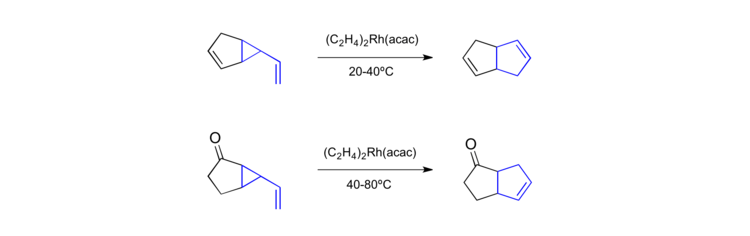
Analogous to the rate acceleration observed in the anionic-oxy-Cope rearrangement Danheiser reported a very similar effect for vinylcyclopropane substrates bearing [alkoxy] substituents.[41]

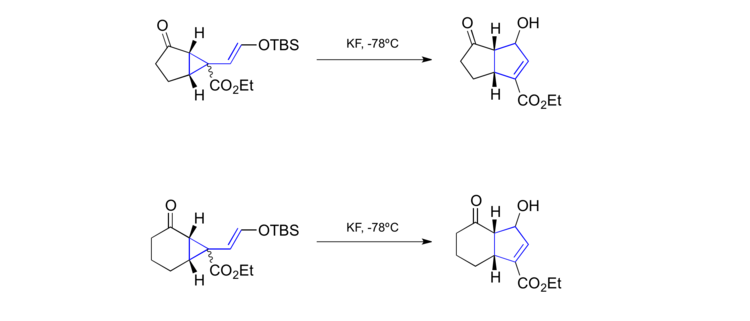
Another intriguing result was reported by Larsen in 1988.[42] He was able to promote vinylcyclopropane rearrangements with substrates such as the one shown in the reaction below at temperatures as low as -78 °C. The substrates were generated in situ upon ringcontracting thiocarbonyl Diels-Alder adducts under basic conditions. This methodology allowed the formation of numerous highly functionalized cyclopentenes in a stereoselective manner.

Another low temperature vinylcyclopropane rearrangement was obtained by the Hudlicky group.[43] The scope of this particular methodology is impressively broad and allows the formation of various [5-5]- as well as [5-6]-carbon scaffolds.
Use in total synthesis
Five-membered carbon rings are ubiquitous structural motifs in natural products. In contrast to the larger, fully "consonant" cyclohexane scaffold cyclopentanes and their derivatives are "dissonant" according to the Lapworth-Evans model of alternating polarities. The dissonance in polarity clearly limits the ways by which cyclopentanes can be disconnected which becomes evident in the decreased number of general methods available for making five-membered rings versus the corresponding six-membered rings. Especially the fact that there is no Diels-Alder-equivalent for the synthesis of five-membered rings has been bothering synthetic chemists for many decades. Consequentially, after the vinylcyclopropane rearrangement was discovered around 1960 it didn't take long for the synthetic community to realize the potential inherent to form cyclopentenes by means of the vinylcyclopropane rearrangement. As the vinylcyclopropane rearrangement progressed as a methodology and the reaction conditions improved during the 1970s, first total syntheses making use of the vinylcycopropane rearrangement started to appear around 1980. Key figures to apply this reaction in total synthesis were Barry M. Trost, Elias J. Corey, Thomas Hudlicky, Leo A. Paquette,
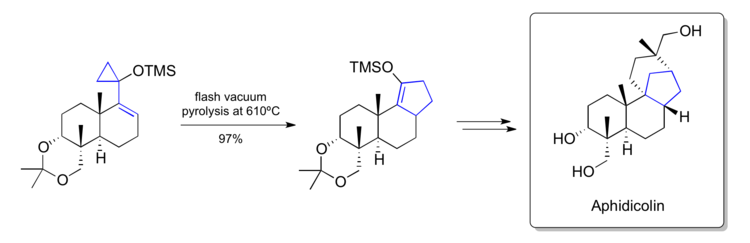
Trost's synthesis of aphidicolin (1979)
In 1979 Trost reported the synthesis of Aphidicolin using methodology around the vinylcyclopropane rearrangement developed in their own laboratory .[44] In one of their key steps they were able to convert a late stage siloxyvinylcyclopropane into a cyclopentene that contained the [6-6-5]-fused carbon skeleton found within the natural product. They were able to convert the rearranged product into the natural product by further manipulations.
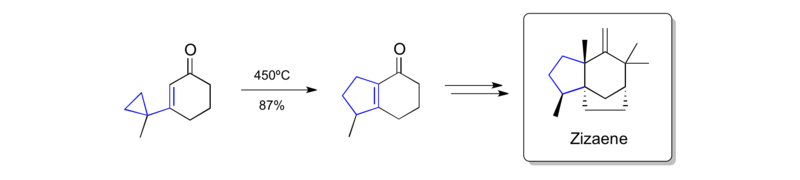
Piers' synthesis of zizaene (1979)
Piers' synthesis of zizaene is another early example for the application of a vinylcyclopropane rearrangement as a key disconnection.[45]
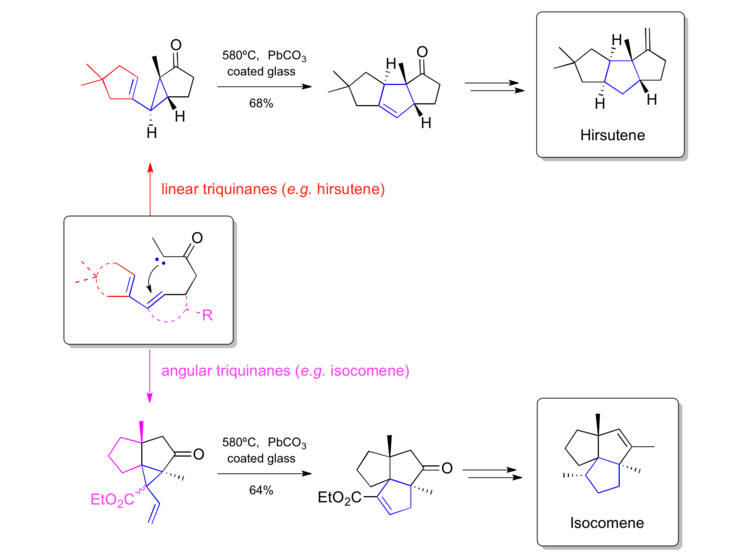
Hudlicky's synthesis of hirstuene (1980) and isocomene (1984)
Hudlicky has been one of the key figures in pushing the vinylcyclopropane rearrangements forwards as a method and has used in multiple times in complex natural product synthesis. A particularly elegant piece of work is the chemistry developed to access both, linear as well as angular triquinanes starting from similar precursors. He has been able to apply this strategy to hirsutene[46] and isocomene[47]
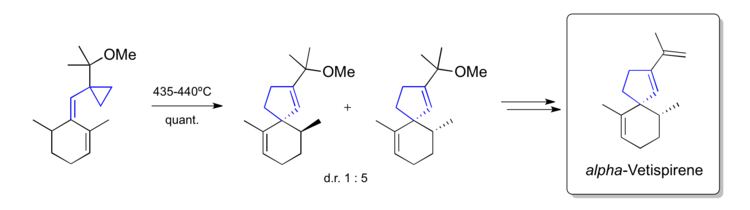
Paquette's synthesis of alpha-Vetispirene (1982)
Paquette used a vinylcyclopropane rearrangement to build the spirocyclic natural product alpha-Vetispirene in 1982.[48]
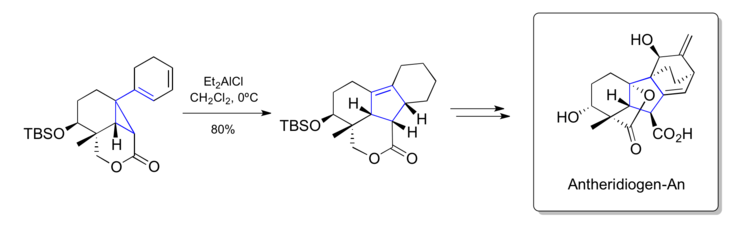
Corey's synthesis of Antheridiogen-An (1985)
Elias J. Corey has contributed heavily to the development of the vinylcyclopropane rearrangement as a synthetic method. In 1985, Corey and his student, Andrew G. Myers, published an impressive synthesis of Antheridiogen-An using a Lewis-acid mediated late-stage vinylcyclopropane rearrangement.[49]
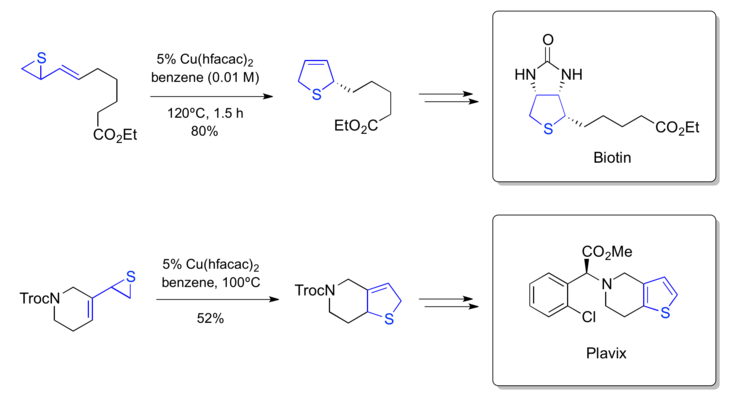
Njardarson's synthesis of biotin (2007)
More recently a copper-catalyzed heteroatom-vinylcyclopropane rearrangement was used to form the tetrahydrothiophene core of biotin and the thiophene unit of Plavix respectively.[50]
Majetich's's synthesis of salviasperanol (2008)
In 2008, an acid-mediated vinylcyclopropane rearrangement was used to synthesize the natural product salviasperanol.[51]

See also
References
- ↑ E M Mil'vitskaya, A V Tarakanova and Alfred F Plate. Thermal Rearrangements of Vinylcyclopropanes. Russ. Chem. Rev. 1976;45:469-478. doi:10.1070/RC1976v045n05ABEH002675.
- ↑ Z. Goldschmidt and B. Crammer. Vinylcyclopropane rearrangements. Chem. Soc. Rev. 1988;17:229-267. doi:10.1039/CS9881700229.
- ↑ Hudlicky, Tomas; Reed, Josephine W. (2010). "From Discovery to Application: 50 Years of the Vinylcyclopropane-Cyclopentene Rearrangement and Its Impact on the Synthesis of Natural Products". Angewandte Chemie International Edition. 49 (29): 4864–76. doi:10.1002/anie.200906001. PMID 20586104.
- ↑ Woodworth, Robert C.; Skell, Philip S. (1957). "Reactions of bivalent carbon species. Addition of dihalocarbenes to 1,3-butadiene". J. Am. Chem. Soc. 79 (10): 2542. doi:10.1021/ja01567a048.
- ↑ Doering, W. von E.; Hoffman, A. Kentaro (1954). "The Addition of Dichlorocarbene to Olefins". J. Am. Chem. Soc. 76 (23): 6162. doi:10.1021/ja01652a087.
- ↑ Neureiter, Norman (1959). "Pyrolysis of 1,l-Dichloro-2-vinylcyclopropane. Synthesis of 2-Chlorocyclopentadiene". J. Org. Chem. 24 (12): 2044. doi:10.1021/jo01094a621.
- ↑ Vogel, Emanuel (1960). "Kleine Kohlenstoff-Ringe". Angewandte Chemie. 72: 4. doi:10.1002/ange.19600720103.
- ↑ Overberger, C. G.; Borchert, A. E. (1960). "Novel thermal rearrangements accompanying acetate pyrolysis in small ring systems". J. Am. Chem. Soc. 82 (4): 1007. doi:10.1021/ja01489a069.
- ↑ Overberger, C. G.; Borchert, A. E. (1960). "Ionic Polymerization. XVI. Reactions of 1-Cyclopropylethanol-Vinylcyclopropane". J. Am. Chem. Soc. 82 (18): 4896. doi:10.1021/ja01503a036.
- ↑ Doering, W. von E.; Lambert, J. B. (1963). "Thermal Reorganization of a- and b-Thujene: A degenerate rearrangement of the vinylcyclopropane type". Tetrahedron. 19 (12): 1989. doi:10.1016/0040-4020(63)85013-9.
- ↑ Atkinson, R. S.; Rees, C. W. (1967). "A vinylaziridine to pyrroline rearrangement". Chemical Communications (London) (23): 1232a. doi:10.1039/C1967001232a.
- ↑ Lwowski, Walter; Rice, Susan N.; Lwowski, Walter (1968). "Singlet and Triplet Nitrenes. 111. The Addition of Carbethoxynitrene to 1,3-Dienes". J. Org. Chem. 33 (22): 481. doi:10.1021/jo01266a001.
- ↑ Paladini, J; Chuche, X. X. (1971). "Rearrangement thermique d'epoxydes vinyliques". Tetrahedron Letters. 12 (46): 4383. doi:10.1016/S0040-4039(01)97447-7.
- ↑ Demjanow, N. J.; Dojarenko, Marie (1922). "Über Vinylcyclopropan, einige Derivate des Methyl-cyclopropyl-carbinols und die Isomerisation des Cyclopropan-Ringes". Ber. Dtsch. Chem. Ges. B. 55 (8): 2718. doi:10.1002/cber.19220550846.
- ↑ Cloke, J. B.; Borchert, A. E. (1929). "The formation of pyrrolines from gamma-chloropropyl and cyclopropyl ketimines". J. Am. Chem. Soc. 51 (18): 1174. doi:10.1021/ja01379a028.
- ↑ Wilson, C. L.; Borchert, A. E. (1947). "Reactions of Furan Compounds. VII. Thermal Interconversionof 2,3-Dihydrofuran and Cyclopropane Aldehyde". J. Am. Chem. Soc. 69 (18): 3002. doi:10.1021/ja01204a020.
- ↑ Baldwin, John E. (2003). "Thermal Rearrangements of Vinylcyclopropanes to Cyclopentenes". Chemical Reviews. 103 (4): 1197–212. doi:10.1021/cr010020z. PMID 12683781.
- ↑ Flowers, M. C.; Rabinovitch, B. S. (1960). "The Thermal Unimolecular Isomerisation of Vinylcyclo- propane to Cyclopentene". J. Chem. Soc. 82 (23): 3547. doi:10.1021/ja01508a008.
- ↑ Schlag, E. W.; Rabinovitch, B. S. (1960). "Kinetics of the Thermal Unimolecular Isomerization Reactions of Cyclopropane-d2". J. Am. Chem. Soc. 82 (23): 5996. doi:10.1021/ja01508a008.
- ↑ Egger, K. W.; Golden, David M.; Benson, Sidney W. (1964). "Iodine-Catalyzed Isomerization of Olefins. 11. The Resonance Energy of the Allyl Radical and the Kinetics of the Positional Isomerization of 1-Butene". J. Am. Chem. Soc. 86 (24): 5420. doi:10.1021/ja01078a011.
- ↑ Woodward, R. B.; Hoffmann, R. (1969). "The Conservation of Orbital Symmetry". Angew. Chem. Int. Ed. 8 (11): 781. doi:10.1002/anie.196907811.
- ↑ Andrews, Gerald D.; Baldwin, John E. (1976). "Stereochemical specificity in the vinylcyclopropane rearrangement vs. MINDO/3 calculations". Journal of the American Chemical Society. 98 (21): 6706. doi:10.1021/ja00437a051.
- ↑ Gajewski, Joseph J.; Squicciarini, Michael P. (1989). "Evidence for concert in the vinylcyclopropane rearrangement. A reinvestigation of the pyrolysis of trans-1-methyl-2-(1-tert-butylethenyl)cyclopropane". Journal of the American Chemical Society. 111 (17): 6717. doi:10.1021/ja00199a035.
- ↑ Baldwin, John E.; Ghatlia, Naresh D. (1991). "Stereochemistry of the thermal isomerization of trans-1-ethenyl-2-methylcyclopropane to 4-methylcyclopentene". Journal of the American Chemical Society. 113 (16): 6273. doi:10.1021/ja00016a055.
- ↑ Gajewski, Joseph J.; Olson, Leif P. (1991). "Evidence for a dominant suprafacial-inversion pathway in the thermal unimolecular vinylcyclopropane to cyclopentene 1,3-sigmatropic shift". Journal of the American Chemical Society. 113 (19): 7432. doi:10.1021/ja00019a056.
- ↑ Baldwin, John E.; Bonacorsi, Samuel (1993). "Stereochemistry of the thermal isomerizations of (1R,2R)-1-[(E)-styryl]-2-methylcyclopropane to 3-phenyl-4-methylcyclopentenes". Journal of the American Chemical Society. 115 (23): 10621. doi:10.1021/ja00076a021.
- ↑ Baldwin, John E.; Villarica, Karla A.; Freedberg, Daron I.; Anet, Frank A. L. (1994). "Stereochemistry of the Thermal Isomerization of Vinylcyclopropane to Cyclopentene". Journal of the American Chemical Society. 116 (23): 10845. doi:10.1021/ja00102a084.
- ↑ Baldwin, John E.; Bonacorsi, Samuel (1994). "Stereochemistry of the Thermal Isomerizations of (1R,2S)-1-(E-1-Propenyl)-2-phenylcyclopropane to 3-Methyl-4-phenylcyclopentenes". The Journal of Organic Chemistry. 59 (24): 7401. doi:10.1021/jo00103a036.
- ↑ Asuncion, Lisa A.; Baldwin, John E. (1995). "Stereochemistry of the Thermal Isomerizations of (1S,2R)-1-(E-Styryl)-2-phenylcyclopropane to 3,4-Diphenylcyclopentenes". Journal of the American Chemical Society. 117 (43): 10672. doi:10.1021/ja00148a009.
- ↑ Asuncion, Lisa A.; Baldwin, John E. (1995). "Quantitative Analyses of the Four Isomers of 3,4-Diphenylcyclopentene by Chiral Gas Chromatography". The Journal of Organic Chemistry. 60 (18): 5778. doi:10.1021/jo00123a010.
- 1 2 Gajewski, Joseph J.; Olson, Leif P.; Willcott, M. Robert (1996). "Evidence for Concert in the Thermal Unimolecular Vinylcyclopropane to Cyclopentene Sigmatropic 1,3-Shift". Journal of the American Chemical Society. 118 (2): 299. doi:10.1021/ja951578p.
- ↑ Houk, K. N.; Nendel, Maja; Wiest, Olaf; Storer, Joey W. (1997). "The Vinylcyclopropane−Cyclopentene Rearrangement: A Prototype Thermal Rearrangement Involving Competing Diradical Concerted and Stepwise Mechanisms". Journal of the American Chemical Society. 119 (43): 10545. doi:10.1021/ja971315q.
- ↑ Corey, E. J.; Walinsky, S. W. (1972). "Reaction of 1,3-dithienium fluoroborate with 1,3-dienes. Synthesis of .DELTA.3-cyclopenten-1-ones". Journal of the American Chemical Society. 94 (25): 8932. doi:10.1021/ja00780a063.
- ↑ Simpson, John M.; Richey, Herman G. (1973). "The effects of methoxyl and phenyl substituents on the thermal rearrangements of vinylcyclopropane". Tetrahedron Letters. 14 (27): 2545. doi:10.1016/S0040-4039(01)96201-X.
- ↑ Trost, Barry M.; Bogdanowicz, Mitchell J. (1973). "New synthetic reactions. IX. Facile synthesis of oxaspiropentanes, versatile synthetic intermediates". Journal of the American Chemical Society. 95 (16): 5311. doi:10.1021/ja00797a036.
- ↑ Trost, Barry M.; Keeley, Donald E. (1976). "New synthetic methods. A stereocontrolled approach to cyclopentane annelation". Journal of the American Chemical Society. 98: 248. doi:10.1021/ja00417a048.
- ↑ Paquette, Leo A.; Meehan, George V.; Henzel, Richard P.; Eizember, Richard F. (1973). "Photochemistry of conjugated cis-bicyclo[5.1.0]octenones, cis- and trans-bicyclo[5.2.0]non-2-en-4-ones, and their methylene analogs". The Journal of Organic Chemistry. 38 (19): 3250. doi:10.1021/jo00959a004.
- ↑ Paquette, Leo A.; Henzel, Richard P.; Eizember, Richard F. (1973). "Thermochemical behavior of conjugated cis-bicyclo[5.1.0]octenones, cis- and trans-bicyclo[5.2.0]non-2-en-4-ones, and their methylene analogs". The Journal of Organic Chemistry. 38 (19): 3257. doi:10.1021/jo00959a005.
- ↑ Hudlicky, Tomas; Koszyk, Francis F.; Kutchan, Toni M.; Sheth, Jagdish P. (1980). "Cyclopentene annulation via intramolecular addition of diazoketones to 1,3-dienes. Applications to the synthesis of cyclopentanoid terpenes". The Journal of Organic Chemistry. 45 (25): 5020. doi:10.1021/jo01313a003.
- ↑ Brown, Vanessa; Brown, John M.; Conneely, John A.; Golding, Bernard T.; Williamson, David H. (1975). "Synthesis and thermolysis of rhodium and iridium complexes of endo-6-vinylbicyclo[3.1.0]hex-2-ene. A metal-promoted vinylcyclopropane to cyclopentene rearrangement". Journal of the Chemical Society, Perkin Transactions 2: 4. doi:10.1039/P29750000004.
- ↑ Danheiser, Rick L.; Martinez-Davila, Carlos; Morin, John M. (1980). "Synthesis of 3-cyclopentenols by alkoxy-accelerated vinylcyclopropane rearrangement". The Journal of Organic Chemistry. 45 (7): 1340. doi:10.1021/jo01295a045.
- ↑ Larsen, Scott D. (1988). "A stereoselective synthesis of functionalized cyclopentenes via base-induced ring contraction of thiocarbonyl Diels-Alder adducts". Journal of the American Chemical Society. 110 (17): 5932. doi:10.1021/ja00225a072.
- ↑ Hudlicky, Tomas; Heard, Nina E.; Fleming, Alison (1990). "4-Siloxy-.alpha.-bromocrotonate: A new reagent for [2+3] annulation leading to oxygenated cyclopentenes at low temperatures". The Journal of Organic Chemistry. 55 (9): 2570. doi:10.1021/jo00296a004.
- ↑ Trost, B. M.; Nishimura, Yoshio; Yamamoto, Kagetoshi (1979). "A Total Synthesis of Aphidicolin". J. Am. Chem. Soc. 101 (5): 1328. doi:10.1021/ja00499a071.
- ↑ Piers, E. (1979). "Five-membered Ring Annelation via Thermal Rearrangement of a-Cyclopropyl-ab-unsaturated Ketones: a New Total Synthesis of (&)-Zizaene". J. Chem. Soc. Chem. Commun.: 1138.
- ↑ Hudlicky, T.; Kutchan, Toni M.; Wilson, Stephen R.; Mao, David T. (1980). "Total Synthesis of (rac)-Hirsutene". J. Am. Chem. Soc. 102 (20): 6351. doi:10.1021/ja00540a036.
- ↑ Hudlicky, T.; Kavka, Misha; Higgs, Leslie A.; Hudlickyl, Tomas (1984). "Stereocontrolled Total Synthesis of Isocomene Sesquiterpenes". Tetrahedron Lett. 25 (23): 2447. doi:10.1016/S0040-4039(01)81201-6.
- ↑ Paquette, L. A. (1982). "A Short Synthesis of (rac)-alpha-Vetispirene". Tetrahedron Lett. 23: 3227. doi:10.1016/s0040-4039(00)87576-0.
- ↑ Corey, E. J.; Myers, Andrew G. (1985). "Total Synthesis of (rac)-Antheridium-Inducing Factor (AAn,2) of the Fern Anemia pbylfitidis. Clarification of Stereochemistry". J. Am. Chem. Soc. 107 (19): 5574. doi:10.1021/ja00305a067.
- ↑ Njardarson, J. T.; Araki, H; Batory, LA; McInnis, CE; Njardarson, JT (2007). "Highly Selective Copper-Catalyzed Ring Expansion of Vinyl Thiiranes: Application to Synthesis of Biotin and the Heterocyclic Core of Plavix". J. Am. Chem. Soc. 129 (10): 2768–9. doi:10.1021/ja069059h. PMID 17302422.
- ↑ Majetich, G.; Zou, G; Grove, J (2008). "Total Synthesis of (−)-Salviasperanol". Org. Lett. 10 (1): 85–7. doi:10.1021/ol701743c. PMID 18052176.