Achondrogenesis
| Achondrogenesis | |
|---|---|
|
The appearance of the female baby with achondrogenesis type I after birth. Baby weighed 1810 grams and measured 31 centimeters; died within the first thirty minutes of birth. | |
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | Q77.0 |
| OMIM | 600972 200610 200600 |
| DiseasesDB | 33398 |
| MedlinePlus | 001247 |

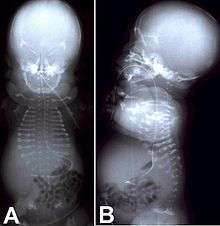
Achondrogenesis is a number of disorders that are the most severe form of congenital chondrodysplasia (malformation of bones and cartilage). These conditions are characterized by a small body, short limbs, and other skeletal abnormalities. As a result of their serious health problems, infants with achondrogenesis are usually born prematurely, are stillborn, or die shortly after birth from respiratory failure. Some infants, however, have lived for a while with intensive medical support.
Researchers have described at least three forms of achondrogenesis, designated as Achondrogenesis type 1A, achondrogenesis type 1B and achondrogenesis type 2. These types are distinguished by their signs and symptoms, inheritance pattern, and genetic cause. Other types of achondrogenesis may exist, but they have not been characterized or their cause is unknown.
Achondrogenesis type 1A is caused by a defect in the microtubules of the Golgi apparatus. In mice, a nonsense mutation in the thyroid hormone receptor interactor 11 gene (Trip11), which encodes the Golgi microtubule-associated protein 210 (GMAP-210), resulted in defects similar to the human disease. When their DNA was sequenced, human patients with achondrogenesis type 1A also had loss-of-function mutations in GMAP-210. GMAP-210 moves proteins from the endoplasmic reticulum to the Golgi apparatus. Because of the defect, GMAP-210 is not able to move the proteins, and they remain in the endoplasmic reticulum, which swells up. The loss of Golgi apparatus function affects some cells, such as those responsible for forming bone and cartilage, more than others.[1]
Achondrogenesis type 1B is caused by a similar mutation in SLC26A2, which encodes a sulfate transporter.
References
- ↑ Lethal skeletal dysplasia in mice and humans lacking the GolginGMAP-210, Patrick Smits et al., N Engl J Med, 362:206, Jan. 21, 2010
This article incorporates public domain text from The U.S. National Library of Medicine
| Wikimedia Commons has media related to Achondrogenesis. |