Birch reduction
| Birch reduction | |
|---|---|
| Named after | Arthur Birch |
| Reaction type | Organic redox reaction |
| Identifiers | |
| Organic Chemistry Portal | birch-reduction |
| RSC ontology ID | RXNO:0000042 |
The Birch reduction is an organic reaction which is particularly useful in synthetic organic chemistry. The reaction was reported in 1944 by the Australian chemist Arthur Birch (1915–1995) working in the Dyson Perrins Laboratory in the University of Oxford,[1][2][3][4][5][6] building on earlier work by Wooster and Godfrey in 1937.[7] It converts aromatic compounds having a benzenoid ring into a product, 1,4-cyclohexadienes, in which two hydrogen atoms have been attached on opposite ends of the molecule. It is the organic reduction of aromatic rings in liquid ammonia with sodium, lithium or potassium and an alcohol, such as ethanol and tert-butanol. This reaction is quite unlike catalytic hydrogenation, which usually reduces the aromatic ring all the way to a cyclohexane.
The original reaction reported by Arthur Birch in 1944 utilized sodium and ethanol.[1][2][3] Subsequently A. L. Wilds noted that better yields result with lithium.[8] Also the use of t-butyl alcohol has become common. The reaction is one of the main organic reactions utilized in all types of syntheses.

An example is the reduction of naphthalene:[9]
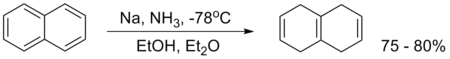
Several reviews have been published.[10][11][12][13]
Basic reaction mechanism
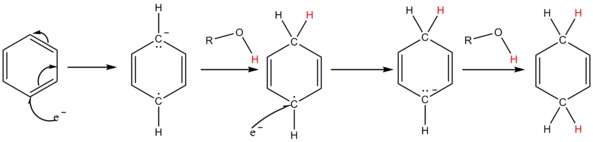
A solution of sodium in liquid ammonia consists of the electride salt [Na(NH3)x]+ e−, associated with the intense blue color of these solutions. The solvated electrons add to the aromatic ring to give a radical anion. The added alcohol supplies a proton to the radical anion and also to the penultimate carbanion; for most substrates ammonia is not acidic enough.[14]
Regioselectivity
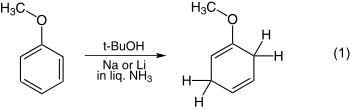
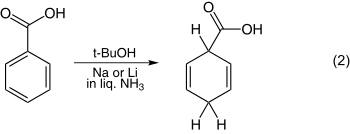
The reduction of anisole is one of the simplest examples and is shown in equation 1. Still another example is that of benzoic acid illustrated in equation 2.
Where the radical-anion is protonated initially determines the structure of the product. With an electron donor such as methoxy (MeO), alkyl protonation has been thought by some investigators as being ortho (i.e. adjacent or 1,2) to the substituent. Other investigators have thought the protonation is meta (1,3) to the substituent. Arthur Birch favored meta protonation. With electron withdrawing substituents protonation has been thought to come at the site (ipso) of the substituent or para (1,4). Again, there has been varied opinion. A. J. Birch’s empirical rules say that for the donor substituents the final product has the maximum number of substituents on the final double bonds. For electron withdrawing groups the double bonds of the product have avoided the substituents. The placement preference of groups in the mechanism and in the final product is termed regioselectivity.
Overall details of the reaction mechanism
The solution of metal in ammonia provides electrons which are taken up by the aromatic ring to form the corresponding radical anion B in the first step of the reaction. This is followed by protonation by the alcohol to form a cyclohexadienyl radical C. Next, a second electron is transferred to the radical to form a cyclohexadienyl carbanion D. In the last step a second proton leads the cyclohexadienyl carbanion to the unconjugated cyclohexadienyl product. These steps are outlined below for the case of anisole.

The reaction is known to be third order – first order in aromatic, first order in the alkali metal, and first order in the alcohol.[15] This requires the rate-limiting step to be the conversion of radical anion B to the cyclohexadienyl radical C.
Reaction regioselectivity
Birch Reduction has several intricate mechanistic features. These features govern the reaction’s regioselectivity and are considered below. Birch’s rule for aromatics with electron donors such as methoxyl or alkyl is that the product will have the residual double bonds bearing the maximum number of substituents. For aromatics with electron withdrawing groups such as carboxyl, the substituent groups avoid the double bonds. In both cases, with electron donating and with withdrawing groups, the residual double bonds are unconjugated (vide infra). It has been a matter of intense interest to understand reaction mechanisms accounting for this regioselectivity. The essential features are:
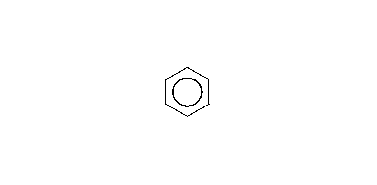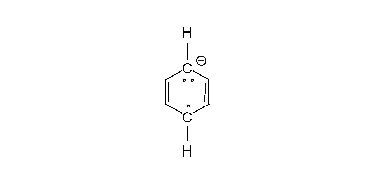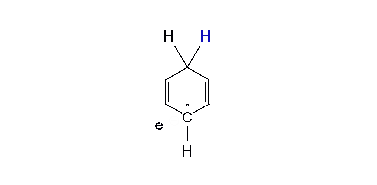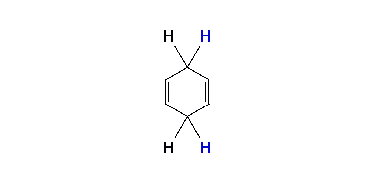
- In liquid ammonia alkali metals dissolve to give a blue solution thought of simplistically as having “free electrons”. The electrons are taken up by the aromatic ring, one at a time. Once the first electron has been absorbed, a radical-anion has been formed. Next the alcohol molecule donates its hydroxylic hydrogen to form a new C-H bond; at this point a radical has been formed. This is followed by the second electron being picked up to give a carbanion of the cyclohexadienyl type (i.e. with C=C-C-C=C in a six-ring and charged minus ). Then this cyclohexadienyl anion is protonated by the alcohol present. The protonation takes place in the middle of the cyclohexadienyl system. This (regio-)selectivity is unique and characteristic.
- Where the radical-anion is protonated initially determines the structure of the product. With an electron donor as methoxy (MeO) or alkyl protonation has been thought by some investigators as being ortho (i.e. adjacent or 1,2) to the substituent. Other investigators have thought the protonation is meta (1,3) to the substituent. Arthur Birch favored meta protonation. With electron withdrawing substituents protonation has been thought to come at the site (ipso) of the substituent or para (1,4). Again, there has been varied opinion. A. J. Birch’s empirical rules say that for the donor substituents the final product has the maximum number of substituents on the final double bonds. For electron withdrawing groups the double bonds of the product have avoided the substituents. The placement preference of groups in the mechanism and in the final product is termed regioselectivity.
- The reaction mechanism provides the details of molecular change as a reaction proceeds. In the case of donating groups A. J. Birch's preference for meta protonation of the radical anion was based on qualitative reasoning. And it had been noted that no experimental test of this was known.
- In 1961 a simple computation of the electron densities of the radical anion revealed that it was the ortho site which was most negative and thus most likely to protonate. However, A. J. Birch seemed to overlook this result. Additionally, the second proton had been determined by the computations to occur in the center of the cyclohexadienyl anion to give an unconjugated product.
- Of historical interest is the uncertainty in the chemical literature at this point. Indeed, there were some further computational results reported. These varied from suggesting a preference for meta radical-anion protonation to suggesting a mixture of ortho and meta protonation.
- In 1990 and 1993 an esoteric test was devised which showed that ortho protonation of the radical anion was preferred over meta (seven to one). This was accompanied by more modern computation which concurred. Both experiment and computations were in agreement with the early 1961 computations.
- With electron withdrawing groups there are literature examples demonstrating the nature of the carbanion just before final protonation. This revealed that the initial radical-anion protonation occurs para to the withdrawing substituent.
- The remaining item for discussion is the final protonation of the cyclohexadienyl anion. In 1961 it was found that simple Hückel computations were unable to distinguish between the different protonation sites. However, when the computations were modified with somewhat more realistic assumptions, the Hückel computations revealed the center carbon to the preferred. The more modern 1990 and 1993 computations were in agreement.
Mechanism
The mechanism of the Birch reduction has been the subject of much discussion. The original mechanism of the Birch reduction invoked protonation of a radical anion that was meta to the ring methoxy and alkyl groups. It further propose that the last step, protonation of a cyclohexadienyl anion, occurred ortho with respect to these substituents. Birch’s original mechanism was based on qualitative reasoning, namely that the radical anion’s electron density, resulting from the addition of an electron, would be highest meta to an electron donor (such as methoxy or methyl) due to avoiding the usual ortho-para high density in the neutral species.[1]
In 1961, simple Hückel computations showed that Birch's proposed mechanism was incorrect. The correct mechanism O is depicted below.[16][17] The two a-priori alternative mechanisms O and M:
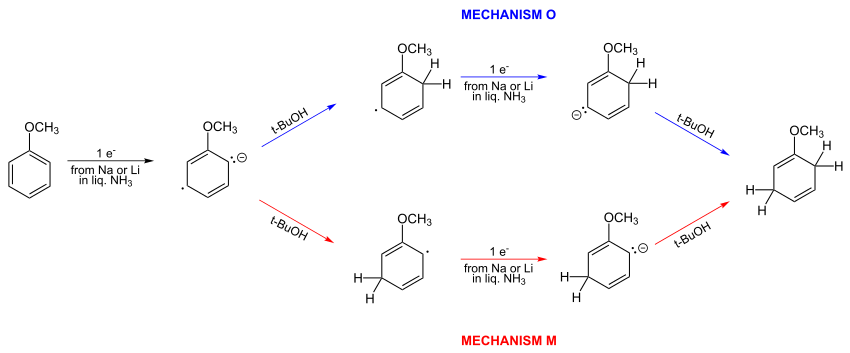
Birch did not accept this conclusion and continued suggesting meta protonation of the radical anion. He suggested the meta attack results from “opposition of the ortho and para initial charge”.[18] Bothner-By in 1959 had given qualitative arguments favoring meta-protonation[15] as had been suggested previously by Birch.
Burnham in 1969 concluded that protonation is unlikely to occur predominantly at the ortho position and the reaction most probably occurs at the meta position but may occur at both sites at similar rates.[19]
Subsequently, Birch, in a review article,[20] noted that no experimental method at the time existed that would distinguish the correct mechanism. But he did note that publication by Burnham[19] favored meta attack.
In 1980 publications Birch collaborated with Leo Radom in a study that concluded that electron densities at the ortho and meta positions to be close with a slight ortho preference, but with mixtures of ortho and meta protonation occurring.[21][22] RHF/sto-3g and UHF/sto-3g computations were used to conclude that both ortho and meta substitutions would occur with a slight preference for ortho.[21][22]
Experimental testing and computational verification
Then in 1990 and 1993 a method was finally devised to experimentally assess whether the anisole and toluene radical anion protonated ortho or meta.[23][24] The esoteric method began with the premise that the isotope selectivity in protonation in a protium–deuterium medium would be greater for the radical anion, of the first protonation step, than for the carbanion of the penultimate step. The reasoning was that carbanions are much more basic than the corresponding radical anions and thus will react more exothermically and less selectively in protonation. Experimentally it was determined that less deuterium at the ortho site than meta resulted (1:7) for a variety of methoxylated aromatics. This is a consequence of the greater selectivity of the radical anion protonation. Computations (e.g. ROHF/6-31g) of the electron densities concurred with the experimental observations. Also, it was ascertained that frontier orbital densities did not, and these had been used in some previous reports.
Subsequently, in 1992 and 1996 Birch published twice still suggesting that meta protonation was preferred.[25][26] This was a reversal of his earlier views as published with Leo Radom.
However, textbooks, publishing on the mechanism of the Birch Reduction, have noted that ortho protonation of the initial radical anion is preferred.[27]
Birch reduction with electron withdrawing substituents
In contrast to the examples with electron donating substituents, the case with withdrawing groups is more readily obvious. Thus, as depicted below, the structure of the penultimate dianion D is characterized by its being subject to trapping by alkyl halides.
Mechanism of reduction of benzoic acids, including possible alkylation

This dianion results independent of whether alcohol is used in the reduction or not. Thus the initial protonation by t-butyl alcohol or ammonia is para rather than ipso as seen in the step from B to C.[28][29][30]
Second step of the Birch reduction with regiochemistry giving unconjugated cyclohexadienes
The second step of the Birch reduction affording unconjugated cyclohexadienes also poses mechanistic questions. Thus as shown in the figure below there are three resonance structures B, C and D for the carbanion. Simple Hückel computations lead, as noted in the first entry of the table below, to equal electron densities at the three atoms 1, 3 and 5. However, in contrast to densities the Hückel computation is less naïve about bond orders,[16][31][32] and bonds 2–3 and 5–6 will be shortened as shown in the first entry of the table. With bond orders modifying simple exchange integrals in a Mulliken-Wheland-Mann computation it was shown that electron density at the central atom 1 become largest.[31][32] More modern RHF computations lead to the same result.[23][24]
Electron introduction to benzene and 3 resonance structures for the carbanion of the second step, and central protonation to give the unconjugated diene:

Five carbons of the cyclohexadienyl anion.[31][32]
| Approximation | Density Atom 3 | Density Atom 2 | Density Atom 1 | Bond Order 2–3 | Bond Order 1–2 |
|---|---|---|---|---|---|
| Hückel (1st approx) | 0.333 | 0.00 | 0.333 | 0.788 | 0.578 |
| 2nd approx | 0.317 | 0.00 | 0.365 | 0.802 | 0.564 |
| 3rd approx | 0.316 | 0.00 | 0.368 | 0.802 | 0.562 |
There are known precedents for central anion protonation.[16][33] Thus conjugated enolates as C=C-C=C-O- have been known for some time as kinetically protonating in the center of the enolate system to afford the β,γ-unsaturated carbonyl compound under conditions where the anion, and not the enol, is the species protonated.
Birch alkylation
In the presence of an alkyl halide the carbanion can also undergo nucleophilic substitution with carbon-carbon bond formation. In substituted aromatic compounds an electron-withdrawing substituent, such as a carboxylic acid,[34] stabilizes a carbanion and the least-substituted olefin is generated. With an electron-donating substituent the opposite effect is obtained.[35] The reaction produces more of the less thermodynamically stable non-conjugated 1,4-addition product than the more stable conjugated 1,3-diene because the largest orbital coefficient of the HOMO of the conjugated pentadienyl anion intermediate is on the central carbon atom. Once formed, the resulting 1,4-cyclohexadiene is unable to equilibrate to the thermodynamically more stable product; therefore, the observed kinetic product is produced. Experimental alkali metal alternatives that are safer to handle, such as the M-SG reducing agent, also exist.
In Birch alkylation the anion formed in the Birch reduction is trapped by a suitable electrophile such as a haloalkane, for example:[36]
In the reaction depicted below, 1,4-dibromobutane is added to t-butyl benzoate to form an alkylated 1,4-cyclohexadiene product:[37]

Modifications of the Birch reduction
Since liquid ammonia has to be condensed into the flask and has to evaporate overnight after the reaction is complete, the whole procedure can be quite troublesome and time-consuming. However, alternative solvents have been employed, such as THF[38][39] as well as a mixture of n-propylamine and ethylenediamine,[40] both with comparable results. The latter one actually is a modification of the Benkeser Reaction, which in its original forms tends to reduce naphthalene all the way to octahydro- and decahydronaphthalene.

This reduction of naphthalene to isotetralin (1,4,5,8-tetrahydronaphthalene) produces some tetralin (1,2,3,4-tetrahydronaphthalene) as byproduct, as is the case with the regular Birch reduction.
See also
References
- 1 2 3 Birch, A. J. (1944). "Reduction by dissolving metals. Part I". J. Chem. Soc.: 430. doi:10.1039/JR9440000430.
- 1 2 Birch, A. J. (1945). "Reduction by dissolving metals. Part II". J. Chem. Soc.: 809. doi:10.1039/jr9450000809.
- 1 2 Birch, A. J. (1946). "Reduction by dissolving metals. Part III". J. Chem. Soc.: 593. doi:10.1039/jr9460000593.
- ↑ Birch, A. J. (1947). "Reduction by dissolving metals. Part IV". J. Chem. Soc.: 102. doi:10.1039/jr9470000102.
- ↑ Birch, Arthur J. (1947). "Reduction by dissolving metals. Part V". J. Chem. Soc.: 1642. doi:10.1039/jr9470001642.
- ↑ Birch, A. J.; Mukherji, S. M. (1949). "Reduction by dissolving metals. Part VI. Some applications in synthesis". J. Chem. Soc.: 2531. doi:10.1039/jr9490002531.
- ↑ Wooster, C. B.; Godfrey, K. L. (1937). "Mechanism of the Reduction of Unsaturated Compounds with Alkali Metals and Water". Journal of the American Chemical Society. 59 (3): 596. doi:10.1021/ja01282a504.
- ↑ Wilds, A. L.; Nelson, N. A. (1953). "A Superior Method for Reducing Phenol Ethers to Dihydro Derivatives and Unsaturated Ketones". J. Am. Chem. Soc. 75 (21): 5360–5365. doi:10.1021/ja01117a064.
- ↑ Vogel, E.; Klug, W.; Breuer, A. (1974). "1,6-Methano[10]annulene". Org. Synth.; Coll. Vol., 6
- ↑ Birch, A. J.; Smith, H. (1958). "Reduction by metal–amine solutions: applications in synthesis and determination of structure". Quart. Rev. (review). 12 (1): 17. doi:10.1039/qr9581200017.
- ↑ Caine, D. (1976). "Reduction and Related Reactions of α,β-Unsaturated Carbonyl Compounds with Metals in Liquid Ammonia". Org. React. (review). 23: 1–258. doi:10.1002/0471264180.or023.01. ISBN 0471264180.
- ↑ Rabideau, P. W.; Marcinow, Z. (1992). "The Birch Reduction of Aromatic Compounds". Org. React. (review). 42: 1–334. doi:10.1002/0471264180.or042.01. ISBN 0471264180.
- ↑ Mander, L. N. (1991). "Partial Reduction of Aromatic Rings by Dissolving Metals and by Other Methods". Comp. Org. Syn. (review). 8: 489–521. doi:10.1016/B978-0-08-052349-1.00237-7. ISBN 978-0-08-052349-1.
- ↑ March, Jerry (1985), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (3rd ed.), New York: Wiley, ISBN 0-471-85472-7
- 1 2 Krapcho, A. P.; Bothner-By, A. A. (1959). "Kinetics of the Metal-Ammonia-Alcohol Reductions of Benzene and Substituted Benzenes1". J. Am. Chem. Soc. 81 (14): 3658–3666. doi:10.1021/ja01523a042.
- 1 2 3 Zimmerman, H. E. (1961). "Orientation in Metal Ammonia Reductions". Tetrahedron. 16: 169–176. doi:10.1016/0040-4020(61)80067-7.
- ↑ "Base-Catalyzed Rearrangements," Chapter 6 of "Molecular Rearrangements," Zimmerman, H. E., Ed. P. DeMayo, Interscience, 345–406, New York, 1963.
- ↑ Birch, A. J.; Nasipuri, D. (1959). "Reaction mechanisms in reduction by metal-ammonia solutions". Tetrahedron. 6 (2): 148–153. doi:10.1016/0040-4020(59)85008-0.
- 1 2 Burnham, D. R. (1969). "Orientation in the mechanism of the Birch reduction of anisole". Tetrahedron. 25 (4): 897–904. doi:10.1016/0040-4020(69)85023-4.
- ↑ Birch, A. J.; Subba Rao, G. (1972). Adv. Org. Chem. 8: 1–65. Missing or empty
|title=(help) (and refs therein) - 1 2 Birch, A. J.; Hinde, A. L.; Radom, L. (1980). "A theoretical approach to the Birch reduction. Structures and stabilities of the radical anions of substituted benzenes". J. Amer. Chem. Soc. 102 (10): 3370–3376. doi:10.1021/ja00530a012.
- 1 2 Birch, A. J.; Radom, L. (1980). "A theoretical approach to the Birch reduction. Structures and stabilities of cyclohexadienyl radicals". J. Amer. Chem. Soc. 102 (12): 4074–4080. doi:10.1021/ja00532a016.
- 1 2 Zimmerman, H. E.; Wang, P. A. (1990). "The Regioselectivity of the Birch Reduction". J. Am. Chem. Soc. 112 (3): 1280–1281. doi:10.1021/ja00159a078.
- 1 2 Zimmerman, H. E.; Wang, P. A. (1993). "Regioselectivity of the Birch Reduction". J. Am. Chem. Soc. 115 (6): 2205–2216. doi:10.1021/ja00059a015.
- ↑ Birch, A. J. (1992). "Steroid hormones and the Luftwaffe. A venture into fundamental strategic research and some of its consequences: The Birch reduction becomes a birth reduction". Steroids. 57 (8): 363–377. doi:10.1016/0039-128X(92)90080-S. PMID 1519267. (showed mechanism w meta)
- ↑ Birch, A. J. (1996). "The Birch reduction in organic synthesis". Pure & Appl. Chem. 68 (3): 553–556. doi:10.1351/pac199668030553. (still suggests meta)
- ↑ “Advanced Organic Chemistry: Reactions and synthesis”, Francis A. Carey, Richard J. Sundberg, p. 437
- ↑ Bachi, J. W.; Epstein, Y.; Herzberg-Minzly, H.; Loewnenthal, J. E. (1969). "Synthesis of compounds related to gibberellic acid. III. Analogs of ring a of the gibberellins". J. Org. Chem. 34: 126–135. doi:10.1021/jo00838a030.
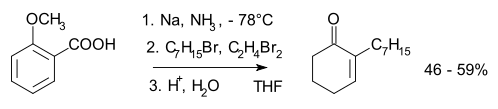
- ↑ Taber, D. F.; Gunn, B.P; Ching Chiu, I (1983). "Alkylation of the Anion from Birch Reduction of o-Anisic Acid: 2-Heptyl-2-Cyclohexenone". Org. Synth. 61: 59.; Coll. Vol., 7, p. 249
- ↑ Guo, Z.; Schultz, A. G. (2001). "Organic synthesis methodology. Preparation and diastereoselective birch reduction-alkylation of 3-substituted 2-methyl-2,3-dihydroisoindol-1-ones". J. Org. Chem. 66 (6): 2154–2157. doi:10.1021/jo005693g. PMID 11300915.
- 1 2 3 Zimmerman, Howard E (1975). Quantum Mechanics for Organic Chemists. New York: Academic Press. pp. 154–5. ISBN 0-12-781650-X.
- 1 2 3 Zimmerman, H. E. in “Molecular Rearrangements”, De Mayo, P. Ed., Interscience, New York, 1963, pp. 350–352
- ↑ Paufler, R. M. (1960) Ph.D. Thesis, Northwestern University, Evanston, IL.
- ↑ Kuehne, M. E.; Lambert, B. F. (1963). "1,4-Dihydrobenzoic acid". Org. Synth.; Coll. Vol., 5, p. 400
- ↑ Paquette, L. A.; Barrett, J. H. (1969). "2,7-Dimethyloxepin". Org. Synth.; Coll. Vol., 5, p. 467
- ↑ Taber, D. F.; Gunn, B. P.; Ching Chiu, I. (1983). "Alkylation of the anion from Birch reduction of o-Anisic acid: 2-Heptyl-2-cyclohexenone". Org. Synth.; Coll. Vol., 7, p. 249
- ↑ Clive, Derrick L. J. & Sunasee, Rajesh (2007). "Formation of Benzo-Fused Carbocycles by Formal Radical Cyclization onto an Aromatic Ring". Organic Letters. 9 (14): 2677–2680. doi:10.1021/ol070849l. PMID 17559217.
- ↑ Ecsery, Zoltan & Muller, Miklos (1961). "Reduction vitamin D2 with alkaly metals". Magyar kémiai folyóirat. 67: 330–332.
- ↑ Donohoe, Timothy J. & House, David (2002). "Ammonia Free Partial Reduction of Aromatic Compounds Using Lithium Di-tert-butylbiphenyl (LiDBB)". Journal of Organic Chemistry. 67 (14): 5015–5018. doi:10.1021/jo0257593. PMID 12098328.
- ↑ Garst, Michael E.; Lloyd J.; Shervin; N. Andrew; Natalie C.; Alfred A.; et al. (2000). "Reductions with Lithium in Low Molecular Weight Amines and Ethylenediamine". Journal of Organic Chemistry. 65 (21): 7098–7104. doi:10.1021/jo0008136. PMID 11031034.