De Vivo disease
| De Vivo disease | |
|---|---|
| Classification and external resources | |
| OMIM | 606777 |
| GeneReviews | |
De Vivo disease is an autosomal dominant developmental disorder associated with a deficiency of GLUT1 also known as Glucose transporter type 1 deficiency syndrome (GLUT1-DS)
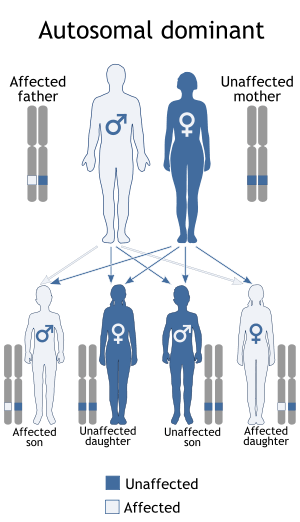
Presentation
De Vivo disease is characterized by deceleration of head growth also known as microcephaly, mental and motor developmental delays, infantile seizures refractory to anticonvulsants, ataxia, dystonia, dysarthria, opsoclonus, spasticity, and other paroxysmal neurologic phenomena. Mothers of infants with this disorder usually have uneventful pregnancies and deliveries, with the child appearing normal and within typical birth weight and length ranges. Infants with GLUT1 deficiency syndrome have a normal head size at birth, but the growth of the brain and skull is slow, in severe cases resulting in an abnormally small head size.[1] Typically, seizures start between one and four months in 90% of cases with abnormal eye movements and apneic episodes preceding the onset of seizures in some cases.[2] Seizures usually are complex to begin with and later become more generalized. Seizure frequency is variable and a history of decreasing frequency during times of ketosis may prompt a diagnosis. Developmental delays are often global and include receptive and expressive language dysfunction.
Genetics
The De Vivo disease is controlled by the SLC2A1,solute carrier family 2 member 1, gene. The SLC2A1 gene's cytogenetic location is 1p34.2.[3] This gene provides the code to produce GLUT1 protein. The GLUT1 protein is used to transport glucose across the blood–brain barrier or the boundary separating tiny blood vessels from brain tissue. Mutations in the SLCA21 gene can cause the glucose transporter to decrease in function or stop completely. As a result, there is less glucose available.[1]
Effects
The disease causes infantile seizures refractory to anticonvulsive drugs, developmental delay, acquired microcephaly and neurologic manifestations including spasticity, hypotonia, and ataxia.[4]
Diagnosis and treatment
De Vivo is diagnosed with CSF glucose value, (<2.2 mmol/L), or lowered CSF/plasma glucose ratio (<0.4), erythrocyte 3-O-methyl-d-glucose uptake assay.[2] Once diagnosed, a Ketogenic diet is usually recommended as it helps to control seizures by providing ketones as an alternative fuel source for the brain other than glucose.[5]
Seeing that the ketogenic diet was discovered as a way to reduce seizures long before 1991 when De Vivo was first diagnosed, it is speculated that some of the ketogentic diet's success was because the children who experienced success on the diet actually had De Vivo at a time when the disease was not yet understood. Less than 100 cases have been identified since its discovery.[1]
References
- 1 2 3 "GLUT1 deficiency syndrome". Genetics Home Reference. Retrieved 10 October 2011.
- 1 2 Wang, Pascual, Vivo. "Glucose Transporter Type 1 Deficiency Syndrome". GeneReviews.
- ↑ "SLC2A1". Genetics Home Reference. Retrieved 6 December 2011.
- ↑ Ticus I, Cano A, Villeneuve N, Milh M, Mancini J, Chabrol B (August 2008). "[GLUT-1 deficiency syndrome or De Vivo disease: a case report]". Arch Pediatr (in French). 15 (8): 1296–9. doi:10.1016/j.arcped.2008.04.024. PMID 18556184.
- ↑ De Vivo, Darryl C.; Trifiletti, Rosario R.; Jacobson, Ronald I.; Ronen, Gabriel M.; Behmand, Ramin A.; Harik, Sami I. (5 September 1991). "Defective Glucose Transport across the Blood-Brain Barrier as a Cause of Persistent Hypoglycorrhachia, Seizures, and Developmental Delay". New England Journal of Medicine. 325 (10): 703–709. doi:10.1056/NEJM199109053251006. PMID 1714544.
External links
- GeneReview/NIH/UW entry on Glucose Transporter Type 1 Deficiency Syndrome
- The Colleen Giblin Research Laboratories for Pediatric Neurology