Galactose-1-phosphate uridylyltransferase deficiency
| Galactose-1-phosphate uridylyltransferase deficiency | |
|---|---|
 | |
| Galactose | |
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E74.2 |
| ICD-9-CM | 271.1 |
| OMIM | 230400 |
| DiseasesDB | 5056 |
| eMedicine | ped/818 |
| MeSH | D005693 |
| GeneReviews | |
Galactose-1-phosphate uridylyltransferase deficiency, also called galactosemia type 1, classic galactosemia or GALT deficiency, is the most common type of galactosemia, an inborn error of galactose metabolism, caused by a deficiency of the enzyme galactose-1-phosphate uridylyltransferase.[1] It is an autosomal recessive metabolic disorder that can cause liver disease and death if untreated. Treatment of galactosemia is most successful if initiated early, and includes dietary restriction of lactose intake. Because early intervention is key, galactosemia is included in newborn screening programs in many areas. On initial screening, which often involves measuring the concentration of galactose in blood, classic galactosemia may be indistinguishable from other inborn errors of galactose metabolism, including galactokinase deficiency and galactose epimerase deficiency. Further analysis of metabolites and enzyme activities are needed to identify the specific metabolic error.
Symptoms and diagnosis
In undiagnosed and untreated children, the accumulation of precursor metabolites due to the deficient activity of galactose 1-phosphate uridylyltransferase (GALT) can lead to feeding problems, failure to thrive, liver damage, bleeding and infections. The first presenting symptom in an infant is often prolonged jaundice. Without intervention in the form of galactose restriction, infants can develop hyperammonemia and sepsis, possibly leading to shock. The accumulation of galactitol and subsequent osmotic swelling can lead to cataracts which are similar to those seen in galactokinase deficiency.[2] Long-term consequences of continued galactose intake can include developmental delay, developmental verbal dyspraxia and motor abnormalities. Galactosemic females frequently suffer from ovarian failure, regardless of treatment in the form of galactose restriction.[2]
In most regions, galactosemia is diagnosed as a result of newborn screening, most commonly by determining the concentration of galactose in a dried blood spot. Some regions will perform a second-tier test of GALT enzyme activity on samples with elevated galactose, while others will immediately initiate confirmatory testing. While awaiting confirmatory testing for classic galactosemia, the infant is typically fed a soy-based formula, as human and cow milk contains galactose.[3] Confirmatory testing would include measurement of enzyme activity in red blood cells, determination of Gal-1-P levels in the blood, and genetic testing. The differential diagnosis for elevated galactose concentrations in blood on a newborn screening result can include other disorders of galactose metabolism, including galactokinase deficiency and galactose epimerase deficiency. Enzyme assays are commonly done using fluorometric detection or radioactively labeled substrates.
Cause
Lactose is a disaccharide consisting of glucose and galactose. After the ingestion of lactose, most commonly from breast milk for an infant or cow milk, the enzyme lactase hydrolyzes the sugar into its monosaccharide constituents, glucose and galactose. In the first step of galactose metabolism, galactose is converted to galactose 1-phosphate (Gal-1-P) by the enzyme galactokinase. Gal-1-P is converted to uridine diphosphate (UDP) galactose by the enzyme galactose-1-phosphate uridylyltransferase, with UDP-glucose acting as a UDP donor. UDP-galactose can then be converted to lactose, by the enzyme lactose synthase, or to UDP-glucose by UDP galactose epimerase.[4]

In classic galactosemia, galactose-1-phosphate uridylyltransferase activity is reduced or absent, leading to an accumulation of the precursors, galactose, galactitol and Gal-1-P.[4] The elevation of precursors can be used to differentiate GALT deficiency from galactokinase deficiency, as Gal-1-P is typically not elevated in galactokinase deficiency. Gal-1-P is assumed as to be a toxic agent, since the inhibition of the Galactokinase prevents toxicity in disease's models.[5][6]
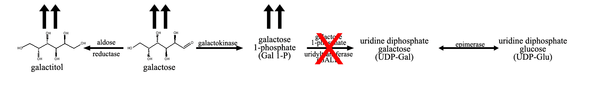
Genetics

All forms of galactosemia are inherited in an autosomal recessive manner, meaning individuals affected with classic galactosemia must have inherited a mutated copy of the GALT gene from both parents. Each child from two carrier parents would have a 25% chance of being affected, a 50% chance of being a carrier, and a 25% chance of inheriting normal versions of the gene from each parent.
There are several variants in the GALT gene, which have different levels of residual enzyme activity. A patient homozygous for the one of the severe mutations in the GALT gene (commonly referred to as G/G) will typically have less than 5% of the enzyme activity expected in an unaffected patient.[2] Duarte galactosemia is caused by mutations that produce an unstable form of the GALT enzyme, with reduced promoter expression. Patients who are homozygous for Duarte mutations (D/D) will have reduced levels of enzyme activity compared to normal controls, but can often maintain a normal diet. Compound heterozygotes (D/G) will often be detected by newborn screening, and treatment is based on the extent of residual enzyme activity.[2]
Treatment
There is no cure for GALT deficiency, in the most severely affected patients, treatment involves a galactose free diet for life. Early identification and implementation of a modified diet greatly improves the outcome for patients. The extent of residual GALT enzyme activity determines the degree of dietary restriction. Patients with higher levels of residual enzyme activity can typically tolerate higher levels of galactose in their diets. As patients get older, dietary restriction is often relaxed.[2] With the increased identification of patients, and their improving outcomes, the management of patients with galactosemia in adulthood is still being understood.
After diagnosis, patients are often supplemented with calcium and vitamin D3. Long-term manifestations of the disease, including ovarian failure in females, ataxia and growth delays are not fully understood.[2] Routine monitoring of patients with GALT deficiency includes determining metabolite levels (galactose 1-phosphate in red blood cells, and galactitol in urine) to measure the effectiveness and adherence of dietary therapy, ophthalmologic examination for the detection of cataracts and assessment of speech, with the possibility of speech therapy if developmental verbal dyspraxia is evident.[2]
References
- ↑ Online Mendelian Inheritance in Man (OMIM) Galactosemia -230400
- 1 2 3 4 5 6 7 Elsas, Louis J (26 October 2010). Galactosemia. PMID 20301691. NBK1518. In Pagon RA, Bird TD, Dolan CR, et al., eds. (1993–). GeneReviews™ [Internet]. Seattle WA: University of Washington, Seattle. Check date values in:
|date=(help) - ↑ "Newborn Screening ACT Sheet [Absent/Reduced Galactose-1-Phosphate Uridyltransferase (GALT)] Classical Galactosemia" (PDF). American College of Medical Genetics. Retrieved 2011-11-05.
- 1 2 Salway, J. G. (2013). "Chart 47.2 Galactose and galactitol metabolism". Metabolism at a Glance (3rd ed.). John Wiley & Sons. p. 102. ISBN 1-118-68207-6.
- ↑ De-Souza, Evandro A.; Pimentel, Felipe S. A.; Machado, Caio M.; Martins, Larissa S.; da-Silva, Wagner S.; Montero-Lomelí, Mónica; Masuda, Claudio A. (2014-01-01). "The unfolded protein response has a protective role in yeast models of classic galactosemia". Disease Models & Mechanisms. 7 (1): 55–61. doi:10.1242/dmm.012641. ISSN 1754-8403.
- ↑ Mumma, Jane Odhiambo; Chhay, Juliet S.; Ross, Kerry L.; Eaton, Jana S.; Newell-Litwa, Karen A.; Fridovich-Keil, Judith L. "Distinct roles of galactose-1P in galactose-mediated growth arrest of yeast deficient in galactose-1P uridylyltransferase (GALT) and UDP-galactose 4′-epimerase (GALE)". Molecular Genetics and Metabolism. 93 (2): 160–171. doi:10.1016/j.ymgme.2007.09.012.