Genotype-first approach
The genotype-first approach is a type of strategy used in genetic epidemiological studies to associate specific genotypes to apparent clinical phenotypes of a complex disease or trait. As opposed to “phenotype-first”, the traditional strategy that have been guiding genome-wide association studies (GWAS) so far, this approach characterizes individuals first by a statistically common genotype based on molecular tests prior to clinical phenotypic classification. This method of grouping leads to patient evaluations based on a shared genetic etiology for the observed phenotypes, regardless of their suspected diagnosis. Thus, this approach can prevent initial phenotypic bias and allow for identification of genes that pose a significant contribution to the disease etiology.[1][2]
This approach is unaffected by phenotypic heterogeneity, incomplete penetrance and levels of expressivity. Therefore, it is useful in complex diseases that also overlap, such as autism spectrum disorder and intellectual disability, enabling the diseases to be distinguished, and specific subtypes of the disease based on the genomic content to be determined.
Currently, the genotype-first approach is used primarily for research objectives. However, the implications from these studies can have valuable clinical applications, including improved diagnosis, counselling, and support groups for individuals with the same genetic etiology.[1]
Background
Initially the idea of identifying the genotype of individuals and subsequently their associated phenotype(s) was first used in early cytogenetic studies. Around 1960 the discovery of Trisomy 21 led to the realization that genetics could be used to predict phenotype(s). From the 1960s to 1990s cytogenetic techniques such as chromosome banding and fluorescence in situ hybridization (FISH) were used to identify and phenotypically characterize patients with chromosomal abnormalities.[3]
Complex diseases and traits pose many difficulties for epidemiological studies due to their nature as multifactorial diseases. More than one gene can underlie a complex disease and generally contributes a smaller effect than what is observed in monogenic diseases (Mendelian diseases). In addition, many of these complex diseases exhibit diverse phenotypes as well as a wide range of expressivity and penetrance. Genes can also be pleiotropic, accounting for many seemingly distinct clinical phenotypes. These features limit the ability of both research and clinical studies to designate causal genes or variants to the observed phenotypes and to classify disorders.
Clinicians are starting to recognize the need to classify genomic diseases by a common genotype rather than a common phenotype and how genotype-first approach can benefit this purpose.[1][4]
Methods
Several methods can be used with a genotype-first approach, however, the following steps are usually included:
- Establishment of a study population and genotyping
- Analysis of genomic variants of interest found in the study population
- Study populations are assembled based on genotype
- Association of genotype to phenotype(s) within respective group
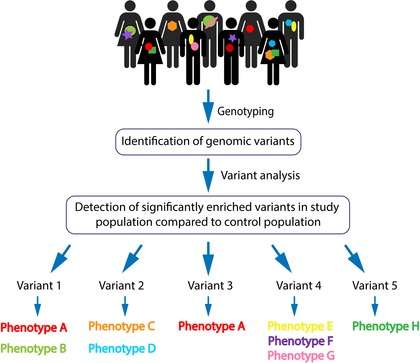
The genotyping is generated using next-generation sequencing technologies (including whole-genome sequencing and exome sequencing) and microarray analyses. The raw data is then statistically analyzed for population-based frequency of the variants. Common variants are filtered out, and pathogenicity is determined though predicted genetic implications. These steps allow for the identification of presumed highly penetrant variants and their specific locus. The selected variants are usually resequenced for validation (by targeted Sanger sequencing). Validated genomic variants can then be analyzed for recurrences among affected individuals within the cohort. Pathogenicity of a genomic variant is statistically based on its significantly abundant presence in the affected compared to the unaffected individuals, not exclusively on the deleteriousness of the variant. A candidate variant can then be associated with a shared phenotype with the aspiration that as more patients baring the same variant with the same phenotype will be identified, a stronger association can be made. Finally, delineation is made between a specific variant to associated clinical phenotypes [Figure 1].
Clinical Implications and Examples
The genotype-first approach has been used to diagnose patients with rare diseases,[5] identify novel disease genotype-phenotypes associations,[6] and characterize uncommon or heterogeneous diseases based on patient's genotype.[1][7] In 2014 the genotype-first approach was used to assess rare and low-frequency variants in the Finnish population. As the Finnish population is isolated and has recently undergone a population bottleneck, relative to other countries, it offers two main benefits for genotype-first studies. Deleterious variants are found at higher frequencies within a smaller spectrum of rare variants in bottlenecked founder populations. By comparing the variants found using whole-exome sequencing (WES) in the Finnish population to WES from a control group of non-Finnish Europeans, loss-of-function (LOF) variants were seen at a higher frequency in the Finnish population. The phenotypes of Finnish individuals with these LOF variants were then analyzed to ascertain novel genotype-phenotype associations. These associations detected included one that could be embryonic lethal, information that might not have been discovered in research using a phenotype-first approach. In addition, researchers also discovered novel splice variants in the LPA gene that reduce apolipoprotein A levels and offer a protective phenotype against cardiovascular disease.[6]
Genotype-first assessment is becoming the standard approach for clinical diagnosis of complex heterogeneous diseases. Microduplication and microdeletion syndromes have a range of characteristics, including intellectual disability and developmental delay, which vary in severity making patients with these syndromes very difficult to diagnose. Since the development of next-generation sequencing technologies, clinicians have been able to use a genotype-first approach to group these patients based on their microdeletion or duplication and document the disease features present in these groups.[8] Chromosomal microarray analysis, in particular, is being used clinically to assist in diagnosing patients with microdeletion and microdulplication syndromes.[4] In diseases, such as Autism spectrum disorder (ASD), where differentiating patients into disease subtype groups based on phenotype is challenging, genotype-first studies allow the classification of patients into subtypes based on their genetics. This in turn will give a greater understanding of the genetic causes of ASD, and could in the future define specific subtypes of ASD for patients to be diagnosed with.[1]
Genotype-first research, through the identification of novel disease-associated genes, can also benefit pharmaceutical companies and drug development. For complex diseases, using phenotype first gene-association, developing therapeutics is often unsuccessful due to multiple genes contributing to one disease. With genotype-first associations, the potential therapeutic target is identified first.[1]
Advantages and Limitations
Advantages
- A shift towards characterizing individuals by a common genotype rather than the clinical presentation will allow for classifying new syndromes and the genetic classification of a certain disease subtypes, as sequencing becoming cheaper, faster and more efficient.[2][9]
- Inheritance of a genomic variant from a healthy parent would not result in its exclusion from variant analysis, thereby accounting for the role of modifiers on phenotypic outcome.[2]
- This approach is unaffected by phenotypic heterogeneity, incomplete penetrance and expressivity.
- This approach contributes to studying both expressivity, pleiotropy and sporadic mutations.
- This approach examines highly penetrant mutations that are associated with the disease regardless of the genetic background.
- Comprehensive and detailed phenotyping is possible even with a small number of patients with common genetic etiology.[1]
- This approach can identify atypical presentations of disease when being used diagnostically.[4]
Limitations
- The phenotype might change over time (e.g. becomes more severe, change in physical location) making genotype-first studies an assumption about the role of the variant in disease manifestation at a specific time point. Therefore, longitudinal follow up is important in order for the genotype-phenotype association to be valued with time and examine the disease’s prognosis.
- Variants identified that might contribute to a mild phenotype, or to range of phenotypes, would not be beneficial in determining diagnosis and prognostic. However, in the future, as more disease subtypes are classified, mild phenotypes could have more relevance.
- Genotype-phenotype association relies on the presentation of clinically recognizable phenotypes.[1]
- As seen in other genome association studies, this approach can generate variants of unknown significance, especially when being used diagnostically.[4]
See also
- Complex disease
- Genome-wide association study (GWAS)
- Microarray
- Whole-exome sequencing (WES)
- Whole-genome sequencing (WGS)
References
- 1 2 3 4 5 6 7 8 Stessman, H. A., Bernier, R. & Eichler, E. E. A genotype-first approach to defining the subtypes of a complex disease. Cell 156, 872–877 (2014).
- 1 2 3 Mefford, H. C. Genotype to phenotype-discovery and characterization of novel genomic disorders in a “genotype-first” era. Genet. Med. 11, 836–842 (2009).
- ↑ Ledbetter, D. H. Cytogenetic Technology — Genotype and Phenotype. N. Engl. J. Med. 359, 1728–1730 (2008).
- 1 2 3 4 Reiff M, Mueller R, Mulchandani S, Spinner NB, Pyeritz RE, Bernhardt B a. A qualitative study of healthcare providers’ perspectives on the implications of genome-wide testing in pediatric clinical practice. J Genet Couns. 2014;23:474–88.
- ↑ Shi, L., Li, B., Huang, Y., Liu, T. & Lyon, G. J. “ Genotype-first ” approaches on a curious case of idiopathic progressive cognitive decline. 1–10 (2014). doi:10.1186/s12920-014-0066-
- 1 2 Lim, E. T. et al. Distribution and Medical Impact of Loss-of-Function Variants in the Finnish Founder Population. PLoS Genet. 10, (2014).
- ↑ Traylor, R. N. et al. A genotype-first approach for the molecular and clinical characterization of uncommon de novo microdeletion of 20q13.33. PLoS One 5, 4–10 (2010).
- ↑ Watson, C. T., Marques-Bonet, T., Sharp, A. J. & Mefford, H. C. The Genetics of Microdeletion and Microduplication Syndromes: An Update. Annu. Rev. Genomics Hum. Genet. 15, 215–244 (2014).
- ↑ Volume 40 of International review of research in developmental disabilities, Deborah J Fidler, Academic Press, 2011)