Inverse electron-demand Diels–Alder reaction

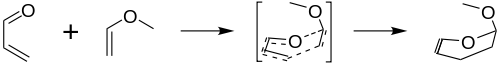
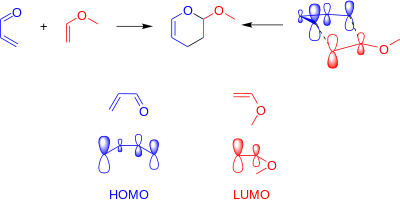
The Inverse electron demand Diels–Alder reaction, or DAINV or IEDDA[1] is an organic chemical reaction, in which two new chemical bonds and a six-membered ring are formed. It is related to the Diels–Alder reaction, but unlike the Diels–Alder (or DA) reaction, the DAINV is a cycloaddition between an electron-rich dienophile and an electron-poor diene.[2] During a DAINV reaction, three pi-bonds are broken, and two sigma bonds and one new pi-bond are formed. A prototypical DAINV reaction is shown on the right.
DAINV reactions often involve heteroatoms, and can be used to form heterocyclic compounds. This makes the DAINV reaction particularly useful in natural product syntheses, where the target compounds often contain heterocycles. Recently, the DAINV reaction has been used to synthesize a drug transport system which targets prostate cancer.[3]
History
The Diels–Alder reaction was first reported in 1928 by Otto Diels and Kurt Alder; they were awarded the Nobel Prize in chemistry for their work in 1950. Since that time, use of the Diels–Alder reaction has become widespread. Conversely, DAINV does not have a clear date of inception, and lacks the comparative notoriety of the standard Diels-Alder. DAINV does not have a clear date of discovery, because of the difficulty that chemists had in differentiating normal from inverse electron-demand Diels-Alder reactions before the advent of modern computational methods.[4] Much of the work in this area is attributed to Dale Boger, though other authors have published numerous papers on the subject.[2][5]
Mechanism
Formal mechanism

The mechanism of the DAINV reaction is controversial. While it is accepted as a formal [4+2] cycloaddition, it is not well understood whether or not the reaction is truly concerted. The accepted view is that most DAINV reactions occur via an asynchronous mechanism. The reaction proceeds via a single transition state, but not all bonds are formed or broken at the same time, as would be the case in a concerted mechanism.[2]
The formal DAINV mechanism for the reaction of acrolein and methyl vinyl ether is shown in the figure to the right. Though not entirely accurate, it provides a useful model for the reaction. During the course of the reaction, three pi-bonds (labeled with red) are broken, and three new bonds are formed (labeled in blue): two sigma bonds and one new pi-bond.[6]
Transition State
Like the standard DA, DAINV reactions proceed via a single boat transition state, despite not being concerted. The single boat transition state is a simplification, but DFT calculations suggest
that the time difference in bond scission and formation is minimal, and that despite potential asynchronicity, the reaction is concerted, with relevant bonds being either partially broken or partially formed at some point during the reaction.[7] The near synchronicity of the DAINV means it can be treated similarly to the standard Diels-Alder reaction.[2]
The reaction can be modeled using a closed, boat-like transition state, with all bonds being in the process of forming or breaking at some given point, and therefore must obey the Woodward–Hoffman general selection rules. This means that for a three component, six electron system, all components must interact in a suprafacial manner (or one suprafacial and two antarfacial). With all components being suprafacial, the allowed transition state is boat-like; a chair-like transition state would result in three two-electron antarafacial components. The chair-like case is thermally disallowed by the Woodward-Hoffman rules.[6]
Molecular orbital theory
Standard DA reactions
In the standard Diels-Alder reaction, there are two components: the diene, which is electron rich, and the dienophile, which is electron poor. The relative electron-richness and electron deficiency of the reactants can best be described visually, in a molecular orbital diagram. In the standard Diels–Alder, the electron rich diene has molecular orbitals that are higher in energy than the orbitals of the electron poor dienophile. This difference in relative orbital energies means that, of the frontier molecular orbitals the HOMO of the diene (HOMOdiene) and the LUMO of the dienophile (LUMOdienophile) are more similar in energy than the HOMOdienophile and the LUMOdiene.[2][8] The strongest orbital interaction is between the most similar frontier molecular orbitals: HOMOdiene and LUMOdienophile.
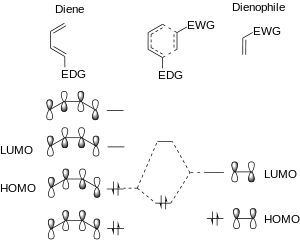
[4+2] dimerization reactions
Dimerization reactions are neither normally or inversely accelerated, and are usually low yielding. In this case, two monomers react in a DA fashion. Because the orbital energies are identical, there is no preference for interaction of the HOMO or the LUMO of either the diene or dienophile. The low yield of dimerization reactions is explained by second-order perturbation theory. The LUMO and HOMO of each species are farther apart in energy in a dimerization than in either normally or inversely accelerated Diels–Alder. This means that the orbitals interact less, and there is a lower thermodynamic drive for dimerization.[2]

Diels–Alder with inverse electron demand
In the dimerization reactions, the diene and dienophile were equally electron rich (or equally electron poor). If the diene becomes any less electron rich, or the dienophile any more so, the possbile [4+2] cycloaddition reaction will then be a DAINV reaction. In the DAINV reaction, the LUMOdiene and HOMOdienophile are closer in energy than the HOMOdiene and LUMOdienophile. Thus, the LUMOdiene and HOMOdienophile are the frontier orbitals that interact the most strongly, and result in the most energetically favourable bond formation.[2][7][9]
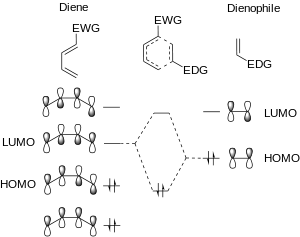
Regiochemistry and stereochemistry of DAINV
Regiochemistry
Regiochemistry in DAINV reactions can be reliably predicted in many cases. This can be done one of two ways, either by electrostatic (charge) control, or orbital control.[2][7][9] To predict the regiochemistry via charge control, one must consider the resonance forms of the reactants. These resonance forms can be used to assign partial charges to each of the atoms. Partially negative atoms on the diene will bond to partially positive atoms on the dienophile, and vice versa.
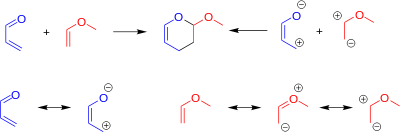
Predicting the regiochemistry of the reaction via orbital control requires one to calculate the relative orbital coefficients on each atom of the reactants.[7] The HOMO of the dienophile reacts with the LUMO of the diene. The relative orbital size on each atom is represented by orbital coefficients in the Frontier molecular orbital theory (FMO). Orbitals will align to maximize the bonding interactions, and minimize the anti-bonding interactions.
Alder–Stein principle
The Alder–Stein principle states that the stereochemistry of the reactants is maintained in the stereochemistry of the products during a Diels–Alder reaction. This means that groups which were cis in relation to one another in the starting materials will be syn to one another in the product, and groups that were trans to one another in the starting material will be anti in the product.
It is important to note that the Alder–Stein principle has no bearing on the relative orientation of groups on the two starting materials. One cannot predict, via this principle, whether a substituent on the diene will be syn or anti to a substituent on the dienophile. The Alder–Stein principle is only consistent across the self-same starting materials. The relationship is only valid for the groups on the diene alone, or the groups on the dienophile, alone. The relative orientation of groups between the two reactants can be predicted by the endo selection rule.
Endo selection rule

Similarly to the standard Diels–Alder reaction, the DAINV also obeys a general endo selection rule. In the standard Diels–Alder, it is known that electron withdrawing groups on the dienophile will approach endo, with respect to the diene. The exact cause of this selectivity is still debated, but the most accepted view is that endo approach maximizes secondary orbital overlap.[10]
The DAINV favors an endo orientation of electron donating substituents on the dienophile. Since all Diels–Alder reactions proceed through a boat transition state, there is an "inside" and an "outside" of the transition state (inside and outside the "boat"). The substituents on the dienophile are considered "endo" if they are 'inside' the boat, and "exo" if they are on the outside.
The exo pathway would be favored by sterics, so a different explanation is needed to justify the general predominance of endo products. Frontier molecular orbital theory can be used to explain this outcome. When the substituents of the dienophile are exo, there is no interaction between those substituents and the diene. However, when the dienophile substituents are endo, there is considerable orbital overlap with the diene. In the case of DAINV the overlap of the orbitals of the electron withdrawing substituents with the orbitals of the diene create a favorable bonding interaction, stabilizing the transition state relative to the exo transition state.[7] The reaction with the lower activation energy will proceed at a greater rate.[7]
Common dienes
The dienes used in Inverse electron demand Diels-Alder are relatively electron-deficient species; compared to the standard Diels-Alder, where the diene is electron rich. These electron-poor species have lower molecular orbital energies than their standard DA counterparts. This lowered energy results from the inclusion of either: A) electron withdrawing group, or B) electronegative heteroatoms. Aromatic compounds can also react in DAINV reactions, such as triazines and tetrazines. Other common classes of dienes are oxo- and aza- butadienes.[9][11]
The key quality of a good DAINV diene is a significantly lowered HOMO and LUMO, as compared to standard DA dienes. Below is a table showing a few commonly used DAINV dienes, their HOMO and LUMO energies, and some standard DA dienes, along with their respective MO energies.[2][12][13][14]
| Diene | Name | HOMO Energy (eV) | LUMO Energy (eV) | Reaction Pathway (DA/DAINV) |
|---|---|---|---|---|
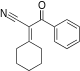 |
2-cyclohexylidene-3-oxo-3-phenylpropanenitrile | -9.558 | 2.38 | DAINV |
| Acrolein | -14.5 | 2.5 | DAINV | |
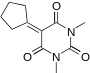 |
5-cyclopentylidene-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione | -10.346 | 1.879 | DAINV |
| Butadiene | -10.346 | 1.879 | DA or DAINV | |
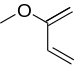 |
1-Methoxy-butadiene | -8.21 | 3.77 | DA |
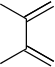 |
2,3-dimethyl-butadiene | -8.76 | 2.18 | DA |
Common dienophiles
The dienophiles used in inverse electron demand Diels-Alder reactions are, unlike in the standard DA, very electron rich, containing one or more electron donating groups. This results in higher orbital energies, and thus more orbital overlap with the LUMO of the diene. Common classes of dieneophiles for DAINV reaction include vinyl ethers and vinyl acetals, imine, enamines, alkynes and highly strained olefins.[11][14]
The most important consideration in choice of dienophile is its relative orbital energies. Both HOMO and LUMO impact the rate and selectivity of the reaction. A table of common DAINV dienophiles, standard DA dienophiles, and their respective MO energies can be seen below.[2][7][12]
| Dienophile | Name | HOMO Energy (eV) | LUMO Energy (eV) | Reaction Pathway (DA/DAINV) |
|---|---|---|---|---|
| ethyl vinyl ether | -9.006 | 5.313 | DAINV | |
 |
2-methylenetetrahydro-2H-pyran | -8.939 | 5.140 | DAINV |
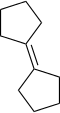 |
1,1'-bis(cyclopentilidene) | -8.242 | 4.749 | DAINV |
| Acrolein | -14.5 | 2.5 | DA | |
 |
Cyclohexene | -8.94 | 2.1 | DA |
| Propene | -9.13 | 1.8 | DA | |
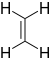 |
Ethylene | -10.52 | 1.5 | DA |
A second table shows how electron richness in the dienophiles affects the rate of reaction with a very electron poor diene, namely hexachlorocyclopentadiene.The more electron rich the dienophile is, the higher the rate of the reaction will be. This is very clear when comparing the relative rates of reaction for styrene and the less electron rich p-nitrostyrene; the more electron rich styrene reactions roughly 40% faster than p-nitrostyrene.[5]
| Dienophile | Relative reaction rate with |
|---|---|
| Cyclopentadiene | 15200 |
| p-Methoxystyrene | 1580 |
| Styrene | 750 |
| p-Nitrostyrene | 538 |
| 2,3-Dihydrofuran | 333 |
| Norbornene | 70.8 |
| Cyclopentene | 59.0 |
| Maleic anhydride | 29.1 |
| Cyclohexene | 3.0 |
Scope and applications
DAINV reactions provide a pathway to a rich library of synthetic targets,[7][11] and have been utilized to form many highly functionalized systems, including selectively protected sugars, an important contribution to the field of sugar chemistry.[15] In addition, DAINV reactions can produce an array of different products from a single starting material, such as tetrazine.[2][13]
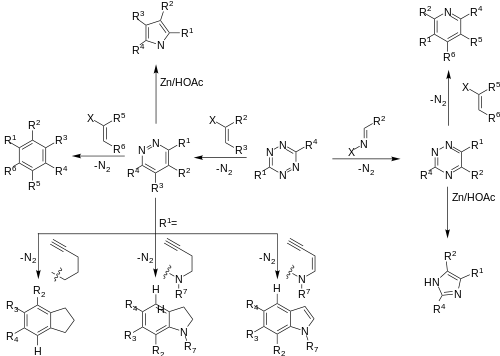
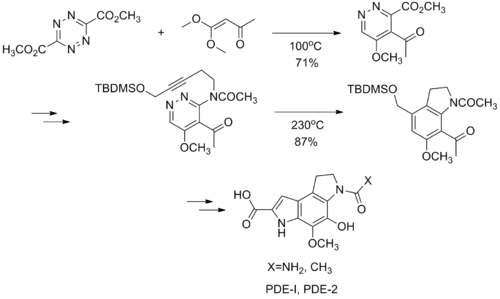
DAINV reactions have been utilized for the synthesis of several natural products, including (-)-CC-1065, a parent compound in the Duocarmycin series, which found use as an anticancer treatment. Several drug candidates in this series have progress into clinical trials. The DAINV reaction was used to synthesise the PDE-I and PDE-II sections of (-)-CC-1065. The first reaction in the sequence is a DAINV reaction between the tetrazine and vinyl acetal, followed by a retro-Diels–Alder reaction to afford a 1,2-diazine product. After several more steps, an intramolecular DAINV reaction occurs, followed again by a retro Diels-Alder in situ, to afford an indoline product. This indoline is a converted into either PDE-I or PDE-II in a few synthetic steps.
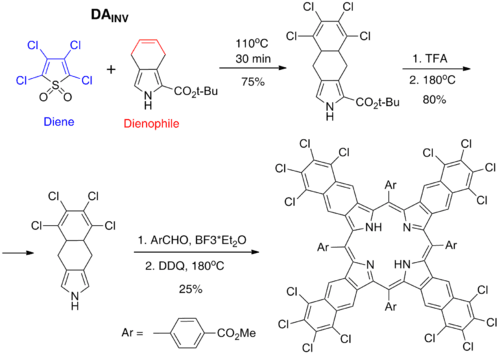
DAINV reaction between 2,3,4,5-tetrachlorothiophene-1,1-dioxide (diene) and 4,7-dihydroisoindole derivative (dienophil) afforded a new precursor for tetranaphthoporphyrins (TNP) bearing perchlorinated aromatic rings. This precursor can be transformed into corresponding porphyrins by Lewis acid-catalyzed condensation with aromatic aldehydes and further oxidation by DDQ. Polychlorination of the TNP system has a profound favorable effect on its solubility. It should be noted that heavy aggregation and poor solubility of the parent tetranaphthoporphyrins severely degrade the usefulness of this potentially very valuable porphyrin family. Thus, the observed effect of polychlorination is very welcome. Besides the effect on the solubility, polychlorination also turned out to improve substantially the stability of these compounds towards photooxidation, which has been known to be another serious drawback of tetranaphthoporphyrins.[16]
See also
- Diels–Alder reaction
- Cycloaddition
- Pericyclic reaction
- Molecular orbital theory
- Boger pyridine synthesis
External links
References
- ↑ Bodwell, Graham J.; Zulan Pi; Ian R. Pottie (1999). "Electron Deficient Dienes. 2. One Step Synthesis of a Coumarin-Fused Electron Deficient Diene and its Inverse Electron Demand Diels–Alder Reactions with Enamines" (PDF). Synlett. 1999 (4): 477–479. doi:10.1055/s-1999-2645. Retrieved 31 March 2013.
- 1 2 3 4 5 6 7 8 9 10 11 Boger, Dale (1989). Progress in heterocyclic chemistry (1st ed.). New York: Pergamon. ISBN 978-0-08-037044-6.
- ↑ Weissler, M (2010). "The Diels-Alder-Reaction with inverse-Electron-Demand, a very efficient versatile Click-Reaction Concept for proper Ligation of variable molecular Partners" (PDF). International Journal of Medical Sciences. 7: 19–28. doi:10.7150/ijms.7.19.
- ↑ Fleischhauer J, Asaad AN, Schleker W, Scharf HD (1981). "Zur Problematik der Einteilung von Diels-Alder-Reaktionen in "normale" und "inverse"" [On the Difficulty of Classifying Diels-Alder-Reactions into “Normal” and “Inverse”]. Liebigs Annalen der Chemie (in German). 1981 (2): 306–311. doi:10.1002/jlac.198119810214. ISSN 0170-2041.
- 1 2 Sauer, J.; Wiest, H. (21 May 1962). "Diels-Alder-Additionen mit "inversem" Elektronenbedarf". Angewandte Chemie. 74 (10): 353–353. doi:10.1002/ange.19620741006.
- 1 2 Woodward, R (1 January 1959). "The mechanism of the Diels-Alder reaction". Tetrahedron. 5 (1): 70–89. doi:10.1016/0040-4020(59)80072-7.
- 1 2 3 4 5 6 7 8 Rooshenas, Parham; Hof, Kira; Schreiner, Peter R.; Williams, Craig M. (1 February 2011). "1,2,4-Triazine vs. 1,3- and 1,4-Oxazinones in Normal- and Inverse-Electron-Demand Hetero-Diels-Alder Reactions: Establishing a Status Quo by Computational Analysis". European Journal of Organic Chemistry. 2011 (5): 983–992. doi:10.1002/ejoc.201001365.
- ↑ Hoffmann, Roald; Woodward, Robert B. (1 January 1968). "Conservation of orbital symmetry". Accounts of Chemical Research. 1 (1): 17–22. doi:10.1021/ar50001a003.
- 1 2 3 Dang, Anh-Thu; Miller, David O.; Dawe, Louise N.; Bodwell, Graham J. (1 January 2008). "Electron-Deficient Dienes. 5. An Inverse-Electron-Demand Diels−Alder Approach to 2-Substituted 4-Methoxyxanthones and 3,4-Dimethoxyxanthones". Organic Letters. 10 (2): 233–236. doi:10.1021/ol702614b.
- ↑ García JI, Mayoral JA, Salvatella L (2005). "The Source of the endoRule in the Diels−Alder Reaction: Are Secondary Orbital Interactions Really Necessary?". European Journal of Organic Chemistry. 2005 (1): 85–90. doi:10.1002/ejoc.200400424. ISSN 1434-193X.
- 1 2 3 Pottie, Ian; Nandaluru, Penchal; Bodwell, Graham (30 August 2011). "An Inverse Electron-Demand Diels-Alder-Based Total Synthesis of Urolithin M7". Synlett. 2011 (15): 2245–2247. doi:10.1055/s-0030-1261203.
- 1 2 Flemming, Ian (2010). Molecular Orbitals and Organic Chemical Reactions. Great Britain: Wiley. ISBN 978-0-470-74658-5.
- 1 2 Figeys, H.P.; Mathy, A. (1 January 1981). "Diels-alder reactions with inverse electron demand. II. The reaction of benzamidine with π-deficient heteroaromatic compounds.". Tetrahedron Letters. 22 (15): 1393–1396. doi:10.1016/S0040-4039(01)90330-2.
- 1 2 Pałasz, Aleksandra; Pałasz, Tadeusz (18 February 2011). "Knoevenagel condensation of cyclic ketones with benzoylacetonitrile and N,N′-dimethylbarbituric acid. Application of sterically hindered condensation products in the synthesis of spiro and dispiropyrans by hetero-Diels–Alder reactions". Tetrahedron. 67 (7): 1422–1431. doi:10.1016/j.tet.2010.12.053.
- ↑ Boger, Dale L.; Robarge, Kirk D. (1 November 1988). "Divergent de novo synthesis of carbohydrates based on an accelerated inverse electron demand Diels-Alder reaction of 1-oxa-1,3-butadienes". The Journal of Organic Chemistry. 53 (24): 5793–5796. doi:10.1021/jo00259a040.
- ↑ Filatov, M.A.; Cheprakov, A. V. (2011). "The synthesis of new tetrabenzo- and tetranaphthoporphyrins via the addition reactions of 4,7-dihydroisoindole". Tetrahedron. 67 (19): 3559–3566. doi:10.1016/j.tet.2011.01.052.