Cycloaddition
A cycloaddition is a pericyclic chemical reaction, in which "two or more unsaturated molecules (or parts of the same molecule) combine with the formation of a cyclic adduct in which there is a net reduction of the bond multiplicity."[1] The resulting reaction is a cyclization reaction. Many but not all cycloadditions are concerted. As a class of addition reaction, cycloadditions permit carbon–carbon bond formation without the use of a nucleophile or electrophile.
Cycloadditions can be described using two systems of notation. An older, but still common, notation is based on the size of linear arrangements of atoms in the reactants. It uses parentheses: (i + j + …) where the variables are the numbers of linear atoms in each reactant. The product is a cycle of size (i + j + …). In this system, the standard Diels-Alder reaction a (4 + 2)cycloaddition, the 1,3-dipolar cycloaddition is a (3 + 2)cycloaddition and cyclopropanation of a carbene with an alkene a (2 + 1)cycloaddition.[1]
A more recent, IUPAC-preferred notation uses square brackets to indicate the number of electrons, rather than carbon atoms, involved in the formation of the product. In the [i + j + …] notation, the standard Diels-Alder reaction is a [4 + 2]cycloaddition, the 1,3-dipolar cycloaddition is [4 + 2].[1]
Thermal cycloadditions and their stereochemistry
Thermal cycloadditions are those cycloadditions where the reactants are in the ground electronic state. They usually have (4n + 2) π electrons participating in the starting material, for some integer n. These reactions occur, for reasons of orbital symmetry, in a suprafacial-suprafacial or antarafacial-antarafacial manner (rare). There are a few examples of thermal cycloadditions which have 4n π electrons (for example the [2 + 2] cycloaddition); these proceed in a suprafacial-antarafacial sense, such as the dimerisation of ketene, in which the orthogonal set of p orbitals allows the reaction to proceed via a crossed transition state.
Photochemical cycloadditions and their stereochemistry
Cycloadditions in which 4n π electrons participate can also occur via photochemical activation. Here, one component has an electron promoted from the HOMO (π bonding) to the LUMO (π* antibonding). Orbital symmetry is then such that the reaction can proceed in a suprafacial-suprafacial manner. An example is the DeMayo reaction. Another example is shown below, the photochemical dimerization of cinnamic acid.[2] The two trans alkenes react head-to-tail, and the isolated isomers are called truxillic acids.
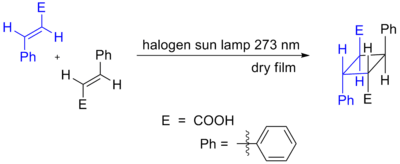
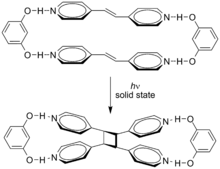
Supramolecular effects can influence these cycloadditions. The cycloaddition of trans-1,2-bis(4-pyridyl)ethene is directed by resorcinol in the solid-state in 100% yield.[3]
Some cycloadditions instead of π bonds operate through strained cyclopropane rings; as these have significant π character. For example, an analog for the Diels-Alder reaction is the quadricyclane-DMAD reaction:

In the (i+j+...) cycloaddition notation i and j refer to the number of atoms involved in the cycloaddition. In this notation a Diels-Alder reaction is a (4+2)cycloaddition and a 1,3-dipolar addition such as the first step in ozonolysis is a (3+2)cycloaddition. The IUPAC preferred notation however, with [i+j+...] takes electrons into account and not atoms. In this notation the DA reaction and the dipolar reaction both become a [4+2]cycloaddition. The reaction between norbornadiene and an activated alkyne is a [2+2+2]cycloaddition.
Types of cycloaddition
Diels-Alder reactions
The Diels-Alder reaction is perhaps the most important and commonly taught cycloaddition reaction. Formally it is a [4+2] cycloaddition reaction and exists in a huge range of forms, including the inverse electron-demand Diels–Alder reaction, Hexadehydro Diels-Alder reaction and the related alkyne trimerisation. The reaction can also be run in reverse in the retro-Diels–Alder reaction.
.png)
Reactions involving heteroatoms are known; including the aza-Diels–Alder and Imine Diels–Alder reaction.
Huisgen cycloadditions
The Huisgen cycloaddition reaction is a (2+3)cycloaddition.

Nitrone-olefin cycloaddition
The Nitrone-olefin cycloaddition is a (3+2)cycloaddition.

Iron-catalyzed 2+2 olefin cycloaddition
Iron[pyridine(diimine)] catalysts contain a redox active ligand in which the central iron atom can coordinate with two simple, unfunctionalized olefin double bonds. The catalyst can be written as a resonance between a structure containing unpaired electrons with the central iron atom in the II oxidation state, and one in which the iron is in the 0 oxidation state. This gives it the flexibility to engage in binding the double bonds as they undergo a cyclization reaction, generating a cyclobutane structure via C-C reductive elimination; alternatively a cyclobutene structure may be produced by beta-hydrogen elimination. Efficiency of the reaction varies substantially depending on the alkenes used, but rational ligand design may permit expansion of the range of reactions that can be catalyzed.[4][5]
Cheletropic reactions
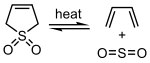
Cheletropic reactions are a subclass of cycloadditions. The key distinguishing feature of cheletropic reactions is that on one of the reagents, both new bonds are being made to the same atom. The classic example is the reaction of sulfur dioxide with a diene.
Other
Other cycloaddition reactions exist: [4+3] cycloadditions, [6+4] cycloadditions, [2+2]photocycloadditions and [4+4] photocycloadditions
Formal cycloadditions
Cycloadditions often have metal-catalyzed and stepwise radical analogs, however these are not strictly speaking pericyclic reactions. When in a cycloaddition charged or radical intermediates are involved or when the cycloaddition result is obtained in a series of reaction steps they are sometimes called formal cycloadditions to make the distinction with true pericyclic cycloadditions.
One example of a formal [3+3]cycloaddition between a cyclic enone and an enamine catalyzed by n-butyllithium is a Stork enamine / 1,2-addition cascade reaction:[6]

References
- 1 2 3 International Union of Pure and Applied Chemistry (IUPAC). "Cycloaddition". Retrieved 26 February 2014.
- ↑ Hein, Sara M. (June 2006). "An Exploration of a Photochemical Pericyclic Reaction Using NMR Data". Journal of Chemical Education. 83 (6): 940–942. Bibcode:2006JChEd..83..940H. doi:10.1021/ed083p940.
- ↑ L. R. MacGillivray; J. L. Reid; J. A. Ripmeester (2000). "Supramolecular Control of Reactivity in the Solid State Using Linear Molecular Templates". J. Am. Chem. Soc. 122 (32): 7817–7818. doi:10.1021/ja001239i.
- ↑ Jordan M. Hoyt; Valeria A. Schmidt; Aaron M. Tondreau; Paul J. Chirik (2015-08-28). "Iron-catalyzed intermolecular [2+2] cycloadditions of unactivated alkenes". Science. 349 (6251): 960–963. doi:10.1126/science.aac7440.
- ↑ Myles W. Smith; Phil S. Baran (2015-08-28). "As simple as [2+2]". Science. 349 (6251): 925–926. doi:10.1126/science.aac9883.
- ↑ Movassaghi, Mohammad; Bin Chen (2007). "Stereoselective Intermolecular Formal [3+3] Cycloaddition Reaction of Cyclic Enamines and Enones". Angew. Chem. Int. Ed. 46 (4): 565–568. doi:10.1002/anie.200603302. PMC 3510678
 . PMID 17146819.
. PMID 17146819.