MT-ND3
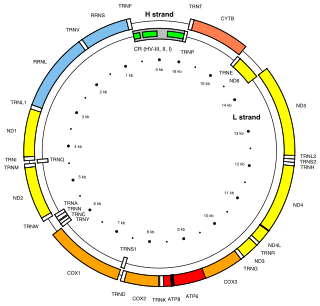
Location of the MT-ND3 gene in the human mitochondrial genome. MT-ND3 is one of the seven NADH dehydrogenase mitochondrial genes (yellow boxes).
Mitochondrially encoded NADH dehydrogenase 3 is a protein that in humans is encoded by the mitochondrial gene MT-ND3.[3] The ND3 protein is a subunit of NADH dehydrogenase (ubiquinone), which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain.[4] Variants of MT-ND3 are associated with Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Leigh's syndrome (LS) and Leber's hereditary optic neuropathy (LHON).[5][6]
Structure
MT-ND3 is located in mitochondrial DNA from base pair 10,059 to 10,404.[3] The MT-ND3 gene produces a 13 kDa protein composed of 115 amino acids.[7][8] MT-ND3 is one of seven mitochondrially-encoded subunits of the enzyme NADH dehydrogenase (ubiquinone). Also known as Complex I, it is the largest of the respiratory complexes. The structure is L-shaped with a long, hydrophobic transmembrane domain and a hydrophilic domain for the peripheral arm that includes all the known redox centres and the NADH binding site. MT-ND3 and the rest of the mitochondrially encoded subunits are the most hydrophobic of the subunits of Complex I and form the core of the transmembrane region.[4]
Function
MT-ND3 is a subunit of the respiratory chain Complex I that is believed to belong to the minimal assembly of core proteins required to catalyze NADH dehydrogenation and electron transfer to ubiquinone (coenzyme Q10).[9] Initially, NADH binds to Complex I and transfers two electrons to the isoalloxazine ring of the flavin mononucleotide (FMN) prosthetic arm to form FMNH2. The electrons are transferred through a series of iron-sulfur (Fe-S) clusters in the prosthetic arm and finally to coenzyme Q10 (CoQ), which is reduced to ubiquinol (CoQH2). The flow of electrons changes the redox state of the protein, resulting in a conformational change and pK shift of the ionizable side chain, which pumps four hydrogen ions out of the mitochondrial matrix.[4]
Clinical significance
Pathogenic variants of the mitochondrial gene MT-ND3 are known to cause mtDNA-associated Leigh syndrome, as are variants of MT-ATP6, MT-TL1, MT-TK, MT-TW, MT-TV, MT-ND1, MT-ND2, MT-ND4, MT-ND5, MT-ND6 and MT-CO3. Abnormalities in mitochondrial energy generation result in neurodegenerative disorders like Leigh syndrome, which is characterized by an onset of symptoms between 12 months and three years of age. The symptoms frequently present themselves following a viral infection and include movement disorders and peripheral neuropathy, as well as hypotonia, spasticity and cerebellar ataxia. Roughly half of affected patients die of respiratory or cardiac failure by the age of three. Leigh syndrome is a maternally inherited disorder and its diagnosis is established through genetic testing of the aforementioned mitochondrial genes, including MT-ND3.[5] These complex I genes have been associated with a variety of neurodegenerative disorders, including Leber's hereditary optic neuropathy (LHON), mitochondrial encephalomyopathy with stroke-like episodes (MELAS) and the previously mentioned Leigh syndrome.[6]
References
- ↑ "Human PubMed Reference:".
- ↑ "Mouse PubMed Reference:".
- 1 2 "Entrez Gene: MT-ND3 NADH dehydrogenase subunit 3".
- 1 2 3 Donald Voet; Judith G. Voet; Charlotte W. Pratt (2013). "18". Fundamentals of biochemistry : life at the molecular level (4th ed.). Hoboken, NJ: Wiley. pp. 581–620. ISBN 9780470547847.
- 1 2 Thorburn DR, Rahman S (1993–2015). "Mitochondrial DNA-Associated Leigh Syndrome and NARP". In Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, Bird TD, Dolan CR, Fong CT, Smith RJ, Stephens K. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle.
- 1 2 La Morgia C, Caporali L, Gandini F, Olivieri A, Toni F, Nassetti S, Brunetto D, Stipa C, Scaduto C, Parmeggiani A, Tonon C, Lodi R, Torroni A, Carelli V. "Association of the mtDNA m.4171C→A/MT-ND1 mutation with both optic neuropathy and bilateral brainstem lesions". BMC Neurology. 14: 116. doi:10.1186/1471-2377-14-116. PMC 4047257
 . PMID 24884847.
. PMID 24884847. - ↑ Zong NC, Li H, Li H, Lam MP, Jimenez RC, Kim CS, Deng N, Kim AK, Choi JH, Zelaya I, Liem D, Meyer D, Odeberg J, Fang C, Lu HJ, Xu T, Weiss J, Duan H, Uhlen M, Yates JR, Apweiler R, Ge J, Hermjakob H, Ping P (Oct 2013). "Integration of cardiac proteome biology and medicine by a specialized knowledgebase". Circulation Research. 113 (9): 1043–53. doi:10.1161/CIRCRESAHA.113.301151. PMC 4076475. PMID 23965338.
- ↑ "NADH-ubiquinone oxidoreductase chain 3". Cardiac Organellar Protein Atlas Knowledgebase (COPaKB).
- ↑ "MT-ND3 - NADH-ubiquinone oxidoreductase chain 3 - Homo sapiens (Human)". UniProt.org: a hub for protein information. The UniProt Consortium.
Further reading
- Torroni A, Achilli A, Macaulay V, Richards M, Bandelt HJ (Jun 2006). "Harvesting the fruit of the human mtDNA tree". Trends in Genetics. 22 (6): 339–45. doi:10.1016/j.tig.2006.04.001. PMID 16678300.
- Horai S, Hayasaka K, Kondo R, Tsugane K, Takahata N (Jan 1995). "Recent African origin of modern humans revealed by complete sequences of hominoid mitochondrial DNAs". Proceedings of the National Academy of Sciences of the United States of America. 92 (2): 532–6. doi:10.1073/pnas.92.2.532. PMC 42775. PMID 7530363.
- Ingman M, Kaessmann H, Pääbo S, Gyllensten U (Dec 2000). "Mitochondrial genome variation and the origin of modern humans". Nature. 408 (6813): 708–13. doi:10.1038/35047064. PMID 11130070.
- Finnilä S, Lehtonen MS, Majamaa K (Jun 2001). "Phylogenetic network for European mtDNA". American Journal of Human Genetics. 68 (6): 1475–84. doi:10.1086/320591. PMC 1226134. PMID 11349229.
- Maca-Meyer N, González AM, Larruga JM, Flores C, Cabrera VM (2003). "Major genomic mitochondrial lineages delineate early human expansions". BMC Genetics. 2: 13. doi:10.1186/1471-2156-2-13. PMC 55343. PMID 11553319.
- Herrnstadt C, Elson JL, Fahy E, Preston G, Turnbull DM, Anderson C, Ghosh SS, Olefsky JM, Beal MF, Davis RE, Howell N (May 2002). "Reduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian, and European haplogroups". American Journal of Human Genetics. 70 (5): 1152–71. doi:10.1086/339933. PMC 447592. PMID 11938495.
- Silva WA, Bonatto SL, Holanda AJ, Ribeiro-Dos-Santos AK, Paixão BM, Goldman GH, Abe-Sandes K, Rodriguez-Delfin L, Barbosa M, Paçó-Larson ML, Petzl-Erler ML, Valente V, Santos SE, Zago MA (Jul 2002). "Mitochondrial genome diversity of Native Americans supports a single early entry of founder populations into America". American Journal of Human Genetics. 71 (1): 187–92. doi:10.1086/341358. PMC 384978. PMID 12022039.
- Sudoyo H, Suryadi H, Lertrit P, Pramoonjago P, Lyrawati D, Marzuki S (2003). "Asian-specific mtDNA backgrounds associated with the primary G11778A mutation of Leber's hereditary optic neuropathy". Journal of Human Genetics. 47 (11): 594–604. doi:10.1007/s100380200091. PMID 12436196.
- Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, Hosseini S, Brandon M, Easley K, Chen E, Brown MD, Sukernik RI, Olckers A, Wallace DC (Jan 2003). "Natural selection shaped regional mtDNA variation in humans". Proceedings of the National Academy of Sciences of the United States of America. 100 (1): 171–6. doi:10.1073/pnas.0136972100. PMC 140917. PMID 12509511.
- Ingman M, Gyllensten U (Jul 2003). "Mitochondrial genome variation and evolutionary history of Australian and New Guinean aborigines". Genome Research. 13 (7): 1600–6. doi:10.1101/gr.686603. PMC 403733. PMID 12840039.
- Kong QP, Yao YG, Sun C, Bandelt HJ, Zhu CL, Zhang YP (Sep 2003). "Phylogeny of east Asian mitochondrial DNA lineages inferred from complete sequences". American Journal of Human Genetics. 73 (3): 671–6. doi:10.1086/377718. PMC 1180693. PMID 12870132.
- Moilanen JS, Finnila S, Majamaa K (Dec 2003). "Lineage-specific selection in human mtDNA: lack of polymorphisms in a segment of MTND5 gene in haplogroup J". Molecular Biology and Evolution. 20 (12): 2132–42. doi:10.1093/molbev/msg230. PMID 12949126.
- Coble MD, Just RS, O'Callaghan JE, Letmanyi IH, Peterson CT, Irwin JA, Parsons TJ (Jun 2004). "Single nucleotide polymorphisms over the entire mtDNA genome that increase the power of forensic testing in Caucasians". International Journal of Legal Medicine. 118 (3): 137–46. doi:10.1007/s00414-004-0427-6. PMID 14760490.
- Crimi M, Papadimitriou A, Galbiati S, Palamidou P, Fortunato F, Bordoni A, Papandreou U, Papadimitriou D, Hadjigeorgiou GM, Drogari E, Bresolin N, Comi GP (May 2004). "A new mitochondrial DNA mutation in ND3 gene causing severe Leigh syndrome with early lethality". Pediatric Research. 55 (5): 842–6. doi:10.1203/01.PDR.0000117844.73436.68. PMID 14764913.
- Tanaka M, Cabrera VM, González AM, Larruga JM, Takeyasu T, Fuku N, Guo LJ, Hirose R, Fujita Y, Kurata M, Shinoda K, Umetsu K, Yamada Y, Oshida Y, Sato Y, Hattori N, Mizuno Y, Arai Y, Hirose N, Ohta S, Ogawa O, Tanaka Y, Kawamori R, Shamoto-Nagai M, Maruyama W, Shimokata H, Suzuki R, Shimodaira H (Oct 2004). "Mitochondrial genome variation in eastern Asia and the peopling of Japan". Genome Research. 14 (10A): 1832–50. doi:10.1101/gr.2286304. PMC 524407. PMID 15466285.
- Palanichamy MG, Sun C, Agrawal S, Bandelt HJ, Kong QP, Khan F, Wang CY, Chaudhuri TK, Palla V, Zhang YP (Dec 2004). "Phylogeny of mitochondrial DNA macrohaplogroup N in India, based on complete sequencing: implications for the peopling of South Asia". American Journal of Human Genetics. 75 (6): 966–78. doi:10.1086/425871. PMC 1182158. PMID 15467980.
- Starikovskaya EB, Sukernik RI, Derbeneva OA, Volodko NV, Ruiz-Pesini E, Torroni A, Brown MD, Lott MT, Hosseini SH, Huoponen K, Wallace DC (Jan 2005). "Mitochondrial DNA diversity in indigenous populations of the southern extent of Siberia, and the origins of Native American haplogroups". Annals of Human Genetics. 69 (Pt 1): 67–89. doi:10.1046/j.1529-8817.2003.00127.x. PMC 3905771. PMID 15638829.
- Achilli A, Rengo C, Battaglia V, Pala M, Olivieri A, Fornarino S, Magri C, Scozzari R, Babudri N, Santachiara-Benerecetti AS, Bandelt HJ, Semino O, Torroni A (May 2005). "Saami and Berbers--an unexpected mitochondrial DNA link". American Journal of Human Genetics. 76 (5): 883–6. doi:10.1086/430073. PMC 1199377. PMID 15791543.
- Rajkumar R, Banerjee J, Gunturi HB, Trivedi R, Kashyap VK (2006). "Phylogeny and antiquity of M macrohaplogroup inferred from complete mt DNA sequence of Indian specific lineages". BMC Evolutionary Biology. 5: 26. doi:10.1186/1471-2148-5-26. PMC 1079809. PMID 15804362.
- Friedlaender J, Schurr T, Gentz F, Koki G, Friedlaender F, Horvat G, Babb P, Cerchio S, Kaestle F, Schanfield M, Deka R, Yanagihara R, Merriwether DA (Jun 2005). "Expanding Southwest Pacific mitochondrial haplogroups P and Q". Molecular Biology and Evolution. 22 (6): 1506–17. doi:10.1093/molbev/msi142. PMID 15814828.
External links