Raman scattering
| Scattering |
|---|
 Feynman diagram of scattering between two electrons by emission of a virtual photon |
Raman scattering or the Raman effect /ˈrɑːmən/ is the inelastic scattering of a photon upon interaction with matter. It was discovered by C. V. Raman and K. S. Krishnan (who was a student of C.V. Raman) in liquids,[1] and independently by Grigory Landsberg and Leonid Mandelstam in crystals.[2] The effect had been predicted theoretically by Adolf Smekal in 1923.[3]
When photons are scattered from an atom or molecule, most photons are elastically scattered (Rayleigh scattering), such that the scattered photons have the same energy (frequency and wavelength) as the incident photons. A small fraction of the scattered photons (approximately 1 in 10 million) are scattered by an excitation, with the scattered photons having a frequency different from, and usually lower than, that of the incident photons.[4] In a gas, Raman scattering can occur with a change in energy of a molecule due to a transition to another (usually higher) energy level. Chemists are primarily concerned with the transitional Raman effect.
History
The inelastic scattering of light was predicted by Adolf Smekal in 1923[3] (and in German-language literature it may be referred to as the Smekal-Raman effect[5]). In 1922, Indian physicist C. V. Raman published his work on the "Molecular Diffraction of Light," the first of a series of investigations with his collaborators that ultimately led to his discovery (on 28 February 1928) of the radiation effect that bears his name. The Raman effect was first reported by C. V. Raman and K. S. Krishnan,[1] and independently by Grigory Landsberg and Leonid Mandelstam, on 21 February 1928 (that is why in the former Soviet Union the priority of Raman was always disputed; thus in Russian scientific literature this effect is usually referred to as "combination scattering" or "combinatory scattering"). Raman received the Nobel Prize in 1930 for his work on the scattering of light.[6]
In 1998 the Raman effect was designated a National Historic Chemical Landmark by the American Chemical Society in recognition of its significance as a tool for analyzing the composition of liquids, gases, and solids.[7]
Description
Degrees of freedom
For any given chemical compound, there are a total of 3N degrees of freedom, where N is the number of atoms in the compound. This number arises from the ability of each atom in a molecule to move in three different directions (x, y, and z).[8] When dealing with molecules, it is more common to consider the movement of the molecule as a whole. Consequently, the 3N degrees of freedom are partitioned into molecular translational, rotational, and vibrational motion. Three of the degrees of freedom correspond to translational motion of the molecule as a whole (along each of the three spatial dimensions). Similarly, three degrees of freedom correspond to rotations of the molecule about the , , and -axes. Linear molecules only have two rotations because rotations along the bond axis do not change the positions of the atoms in the molecule. The remaining degrees of freedom correspond to molecular vibrational modes. These modes include stretching and bending motions of the chemical bonds of the molecule. For a linear molecule, the number of vibrational modes is:[8]




whereas for a non-linear molecule the number of vibrational modes are

Molecular vibrations and infrared radiation
The frequencies of molecular vibrations range from less than 1012 to approximately 1014 Hz. These frequencies correspond to radiation in the infrared (IR) region of the electromagnetic spectrum. At any given instant, each molecule in a sample has a certain amount of vibrational energy. However, the amount of vibrational energy that a molecule has continually changes due to collisions and other interactions with other molecules in the sample.
At room temperature, most of the molecules will be in the lowest energy state, which is known as the ground state. A few molecules will be in higher energy states, which are known as excited states. The fraction of molecules occupying a given vibrational mode at a given temperature can be calculated using the Boltzmann distribution. Performing such a calculation shows that, for relatively low temperatures (such as those used for most routine spectroscopy), most of the molecules occupy the ground vibrational state. Such a molecule can be excited to a higher vibrational mode through the direct absorption of a photon of the appropriate energy. This is the mechanism by which IR spectroscopy operates: infrared radiation is passed through the sample, and the intensity of the transmitted light is compared with that of the incident light. A reduction in intensity at a given wavelength of light indicates the absorption of energy by a vibrational transition. The energy, , of a photon is

,

where is Planck’s constant and is the frequency of the radiation. Thus, the energy required for such a transition may be calculated if the frequency of the incident radiation is known.


Raman scattering
It is also possible to observe molecular vibrations by an inelastic scattering process. In inelastic (Raman) scattering, an absorbed photon is re-emitted with lower energy; the difference in energy between the incident photons and scattered photons corresponds to the energy required to excite a molecule to a higher vibrational mode.
Typically, in Raman spectroscopy high intensity laser radiation with wavelengths in either the visible or near-infrared regions of the spectrum is passed through a sample. Photons from the laser beam produce an oscillating polarization in the molecules, exciting them to a virtual energy state. The oscillating polarization of the molecule can couple with other possible polarizations of the molecule, including vibrational and electronic excitations. If the polarization in the molecule does not couple to these other possible polarizations, then it will not change the vibrational state that the molecule started in and the scattered photon will have the same energy as the original photon. This type of scattering is known as Rayleigh scattering.
When the polarization in the molecules couples to a vibrational state that is higher in energy than the state they started in, then the original photon and the scattered photon differ in energy by the amount required to vibrationally excite the molecule. In perturbation theory, the Raman effect corresponds to the absorption and subsequent emission of a photon via an intermediate quantum state of a material. The intermediate state can be either a "real", i.e., stationary state or a virtual state.
Stokes and anti-Stokes scattering
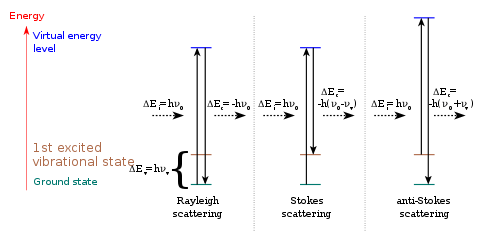
The Raman interaction leads to two possible outcomes:
- the material absorbs energy and the emitted photon has a lower energy than the absorbed photon. This outcome is labeled Stokes Raman scattering in honor of George Stokes who showed in 1852 that fluorescence is due to light emission at longer wavelength (now known to correspond to lower energy) than the absorbed incident light.
- the material loses energy and the emitted photon has a higher energy than the absorbed photon. This outcome is labeled anti-Stokes Raman scattering.
The energy difference between the absorbed and emitted photon corresponds to the energy difference between two resonant states of the material and is independent of the absolute energy of the photon.
The spectrum of the scattered photons is termed the Raman spectrum. It shows the intensity of the scattered light as a function of its frequency difference Δν to the incident photons. The locations of corresponding Stokes and anti-Stokes peaks form a symmetric pattern around Δν=0. The frequency shifts are symmetric because they correspond to the energy difference between the same upper and lower resonant states. The intensities of the pairs of features will typically differ, though. They depend on the populations of the initial states of the material, which in turn depend on the temperature. In thermodynamic equilibrium, the upper state will be less populated than the lower state. Therefore, the rate of transitions from the lower to the upper state (Stokes transitions) will be higher than in the opposite direction (anti-Stokes transitions). Correspondingly, Stokes scattering peaks are stronger than anti-Stokes scattering peaks. Their ratio depends on the temperature, and can therefore be exploited to measure it.
Distinction from fluorescence
The Raman effect differs from the process of fluorescence in that it is a scattering process. For fluorescence, the incident light is completely absorbed, transferring the system to an excited state. After a certain resonance lifetime, the system de-excites to lower energy states via emission of photons. The result of both processes is in essence the same: A photon with a frequency different from that of the incident photon is produced and the molecule is brought to a higher or lower energy level. But the major difference is that the Raman effect can take place for any frequency of incident light. In contrast to the fluorescence effect, the Raman effect is therefore not a resonant effect. In practice, this means that a fluorescence peak is anchored at a specific frequency, whereas a Raman peak maintains a constant separation from the excitation frequency.
Selection rules
A Raman transition from one state to another is allowed only if the molecular polarizability of those states is different. For a vibration, this means that the derivative of the polarizability with respect to the normal coordinate associated to the vibration is non zero: . In general, a normal mode is Raman active if it transforms with the same symmetry of the quadratic forms (), which can be verified from the character table of the molecule's symmetry group.


The specific selection rules state that the allowed rotational transitions are , where is the rotational state.


The allowed vibrational transitions are , where is the vibrational state.

Stimulated Raman scattering and Raman amplification
The Raman-scattering process as described above takes place spontaneously; i.e., in random time intervals, one of the many incoming photons is scattered by the material. This process is thus called spontaneous Raman scattering.
On the other hand, stimulated Raman scattering can take place when some Stokes photons have previously been generated by spontaneous Raman scattering (and somehow forced to remain in the material), or when deliberately injecting Stokes photons ("signal light") together with the original light ("pump light"). In that case, the total Raman-scattering rate is increased beyond that of spontaneous Raman scattering: pump photons are converted more rapidly into additional Stokes photons. The more Stokes photons are already present, the faster more of them are added. Effectively, this amplifies the Stokes light in the presence of the pump light, which is exploited in Raman amplifiers and Raman lasers.
Stimulated Raman scattering is a nonlinear-optical effect. It can be described using a third-order nonlinear susceptibility .

Need of space-coherence
Suppose that the distance between two points A and B of an exciting beam is x. Generally, as the exciting frequency is not equal to the scattered Raman frequency, the corresponding relative wavelengths λ and λ' are not equal. Thus, a phase-shift Θ = 2πx(1/λ − 1/λ') appears. For Θ = π, the scattered amplitudes are opposite, so that the Raman scattered beam remains weak.
- A crossing of the beams may limit the path x.
Several tricks may be used to get a larger amplitude:
- In an optically anisotropic crystal, a light ray may have two modes of propagation with different polarizations and different indices of refraction. If energy may be transferred between these modes by a quadrupolar (Raman) resonance, phases remain coherent along the whole path, transfer of energy may be large. It is an Optical parametric generation.
- Light may be pulsed, so that beats do not appear.
It is the Impulsive Stimulated Raman Scattering (ISRS),[9][10][11][12][13] in which the length of the pulses must be shorter than all relevant time constants.[14] Interference of Raman and incident lights is too short to allow beats, so that it produces a frequency shift roughly, in best conditions, inversely proportional to cube of length of pulses. In labs, femtosecond laser pulses must be used because the ISRS becomes very weak if the pulses are too long. Thus ISRS cannot be observed using nanosecond pulses making ordinary time-incoherent light.
Applications
Raman spectroscopy employs the Raman effect for substances analysis. The spectrum of the Raman-scattered light depends on the molecular constituents present and their state, allowing the spectrum to be used for material identification and analysis. Raman spectroscopy is used to analyze a wide range of materials, including gases, liquids, and solids. Highly complex materials such as biological organisms and human tissue[15] can also be analyzed by Raman spectroscopy.
For solid materials, Raman scattering is used as a tool to detect high-frequency phonon and magnon excitations.
Raman lidar is used in atmospheric physics to measure the atmospheric extinction coefficient and the water vapour vertical distribution.
Stimulated Raman transitions are also widely used for manipulating a trapped ion's energy levels, and thus basis qubit states.
Raman spectroscopy can be used to determine the force constant and bond length for molecules that do not have an infrared absorption spectrum.
Raman amplification is used in optical amplifiers.
Supercontinuum generation
For high-intensity continuous wave (CW) lasers, SRS can be used to produce broad bandwidth spectra. This process can also be seen as a special case of four-wave mixing, wherein the frequencies of the two incident photons are equal and the emitted spectra are found in two bands separated from the incident light by the phonon energies. The initial Raman spectrum is built up with spontaneous emission and is amplified later on. At high pumping levels in long fibers, higher-order Raman spectra can be generated by using the Raman spectrum as a new starting point, thereby building a chain of new spectra with decreasing amplitude. The disadvantage of intrinsic noise due to the initial spontaneous process can be overcome by seeding a spectrum at the beginning, or even using a feedback loop as in a resonator to stabilize the process. Since this technology easily fits into the fast evolving fiber laser field and there is demand for transversal coherent high-intensity light sources (i.e., broadband telecommunication, imaging applications), Raman amplification and spectrum generation might be widely used in the near-future.
See also
- Scattering
- Brillouin scattering
- Nonlinear optics
- Fiber amplifier
- List of surface analysis methods
- Raman laser
- Raman spectroscopy
- Surface Enhanced Raman Spectroscopy (SERS)
- Inverse Raman effect
- Resonance Raman spectroscopy (RR)
- Coherent anti-Stokes Raman spectroscopy (CARS)
- Depolarization ratio
References
- 1 2 Raman, C. V. (1928). "A new radiation". Indian J. Phys. 2: 387–398. Retrieved 14 April 2013.
- ↑ Landsberg, G.; Mandelstam, L. (1928). "Eine neue Erscheinung bei der Lichtzerstreuung in Krystallen". Naturwissenschaften. 16 (28): 557. Bibcode:1928NW.....16..557.. doi:10.1007/BF01506807.
- 1 2 Smekal, A. (1923). "Zur Quantentheorie der Dispersion". Naturwissenschaften. 11 (43): 873–875. Bibcode:1923NW.....11..873S. doi:10.1007/BF01576902.
- ↑ Harris and Bertolucci (1989). Symmetry and Spectroscopy. Dover Publications. ISBN 0-486-66144-X.
- ↑ Nature (1931-12-19). "A review of the 1931 book ''Der Smekal-Raman-Effekt''". Nature.com. Retrieved 2011-09-17.
- ↑ Singh, R. (2002). "C. V. Raman and the Discovery of the Raman Effect". Physics in Perspective. 4 (4): 399–420. Bibcode:2002PhP.....4..399S. doi:10.1007/s000160200002.
- ↑ "C. V. Raman: The Raman Effect". American Chemical Society. Retrieved June 6, 2012.
- 1 2 Keith J. Laidler and John H. Meiser, Physical Chemistry (Benjamin/Cummings 1982), p.646-7 ISBN 0-8053-5682-7
- ↑ A. M. Weiner, D. E. Leaird, Gary P. Wiederrecht, and Keith A. Nelson "Femtosecond multiple-pulse impulsive stimulated Raman scattering spectroscopy" JOSA B, Vol. 8, Issue 6, pp. 1264-1275 (1991)
- ↑ L. Dhar, J. A. Rogers, and K. A. Nelson, “Time-resolved vibrational spectroscopy in the impulsive limit”, Chem. Rev., 1994, 94, 157-193.
- ↑ S. De Silvestri, J.G. Fujimoto, E.P. Ippen, Edward B. Gamble Jr.3, Leah Ruby Williams4, Keith A. Nelson, Femtosecond time-resolved measurements of optic phonon dephasing by impulsive stimulated raman scattering in α-perylene crystal from 20 to 300 K, Chemical Physics Letters, Volume 116, Issues 2–3, Pages 146–152, 1985
- ↑ R. Kosloff, A. D. Hammerich, D. Tannor Excitation without demolition: Radiative excitation of ground-surface vibration by impulsive stimulated Raman scattering with damage control. phys. rev. letters, 69, 15, p. 2172-2175
- ↑ P Voehringer, NF Scherer -Transient grating optical heterodyne detected impulsive stimulated Raman scattering in simple liquids, The Journal of Physical Chemistry, 99 p.2684-96 1995
- ↑ G. L. Lamb Jr., “Analytical description of ultra-short optical pulse propagation in a resonant medium”, Rev. Mod. Phys., 1971, 43, 99-124.
- ↑ "Painless laser device could spot early signs of disease". BBC News. 27 September 2010.
External links
- Explanation from Hyperphysics in Astronomy section of gsu.edu
- Raman Spectroscopy - Tutorial at Kosi.com
- Prof. R. W. Wood Demonstrating the New "Raman Effect" in Physics (Scientific American, December 1930)
- A short description of spontaneous Raman scattering
- Raman Effect: fingerprinting the universe