Reuptake
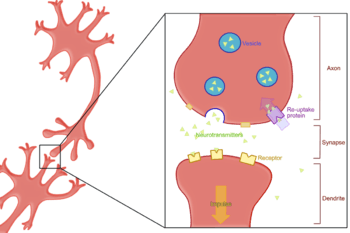
Reuptake, or re-uptake, is the reabsorption of a neurotransmitter by a neurotransmitter transporter of a pre-synaptic neuron after it has performed its function of transmitting a neural impulse.
Reuptake is necessary for normal synaptic physiology because it allows for the recycling of neurotransmitters and regulates the level of neurotransmitter present in the synapse, thereby controlling how long a signal resulting from neurotransmitter release lasts. Because neurotransmitters are too large and hydrophilic to diffuse through the membrane, specific transport proteins are necessary for the reabsorption of neurotransmitters. Much research, both biochemical and structural, has been performed to obtain clues about the mechanism of reuptake.
Protein structure
The first primary sequence of a reuptake protein was published in 1990. The technique for protein sequence determination relied upon the purification, sequencing, and cloning of the transporter protein in question, or expression cloning strategies in which transport function was used as an assay for cDNA species coding for that transporter. After separation, it was realized that there were many similarities between the two DNA sequences. Further exploration in the field of reuptake proteins found that many of the transporters associated with important neurotransmitters within the body were also very similar in sequence to the GABA and norepinephrine transporters. The members of this new family include transporters for dopamine, norepinephrine, serotonin, glycine, proline and GABA. They were called Na+/Cl− dependent neurotransmitter transporters. Sodium and Chloride ion dependence will be discussed later in the mechanism of action. Using the commonalities among sequences and hydropathy plot analyses, it was predicted that there are 12 hydrophobic membrane spanning regions in the ‘Classical’ transporter family.[1] In addition to this, the N- and C-termini exist in the intracellular space. These proteins also all have an extended extracellular loop between the third and fourth transmembrane sequences. Site-directed chemical labeling experiments verified the predicted topological organization of the serotonin transporter.[2]
In addition to neurotransmitter transporters, many other proteins in both animals and prokaryotes were found with similar sequences, indicating a larger family of Neurotransmitter:Sodium Symporters (NSS). One of these proteins, LeuT, from Aquifex aeolicus, was crystallized by Yamashita et al.[3] with very high resolution, revealing a molecule of leucine and two Na+ ions bound near the center of the protein. They found that the transmembrane (TM) helices 1 and 6 contained unwound segments in the middle of the membrane. Along with these two helices, TM helices 3 and 8 and the areas surrounding the unwound sections of 1 and 6 formed the substrate and sodium ion binding sites. The crystal structure revealed pseudo-symmetry in LeuT, in which the structure of TM helices 1-5 is reflected in the structure of helices 6-10.
There is an extracellular cavity in the protein, into which protrudes a helical hairpin formed by extracellular loop EL4. In TM1, an aspartate distinguishes monoamine NSS transporters from amino acid transporters which contain a glycine at the same position. External and internal “gates” were assigned to pairs of negatively and positively charged residues in the extracellular cavity and near the cytoplasmic ends of TM helices 1 and 8.
Mechanism of action
The classic transporter proteins use transmembrane ion gradients and electrical potential to transport neurotransmitter across the membrane of the presynaptic neuron. Typical NSS neurotransmitter transporters, which are Na+ and Cl− ion dependent, take advantage of both Na+ and Cl− gradients, inwardly directed across the membrane. The ions flow down their concentration gradients, in many cases leading to transmembrane charge movement that is enhanced by the membrane potential. These forces pull the neurotransmitter substrate into the cell, even against its own concentration gradient. At a molecular level, Na+ ions stabilize amino acid binding at the substrate site and also hold the transporter in an outward-open conformation that allows substrate binding.[4] The role of the Cl− ion in the symport mechanism has been proposed to be for stabilizing the charge of the symported Na+.[5][6]
After ion and substrate binding have taken place, some conformational change must occur. From the conformational differences between the structure of TMs 1-5 and that of TMs 6-10, and from the identification of a substrate permeation pathway between the binding site of SERT and the cytoplasm, a mechanism for conformational change was proposed in which a four-helix bundle composed of TMs 1, 2, 6 and 7 changes its orientation within the rest of the protein.[7] A structure of LeuT in the inward-open conformation subsequently demonstrated that the major component of the conformational change represents movement of the bundle relative to the rest of the protein.[8]
Mechanism of reuptake inhibition
The main objective of a reuptake inhibitor is to substantially decrease the rate by which neurotransmitters are reabsorbed into the presynaptic neuron, increasing the concentration of neurotransmitter in the synapse. This increases neurotransmitter binding to pre- and postsynaptic neurotransmitter receptors. Depending on the neuronal system in question, a reuptake inhibitor can have drastic effects on cognition and behavior. Non-competitive inhibition of the bacterial homologue LeuT by tricyclic antidepressants resulted from binding of these inhibitors in the extracellular permeation pathway.[9][10] However, the competitive nature of serotonin transport inhibition by antidepressants suggests that in neurotransmitter transporters, they bind in a site overlapping the substrate site.[11]
Human systems
Horschitz et al.[12] examined reuptake inhibitor selectivity among the rat serotonin reuptake protein (SERT) expressed in human embryonic kidney cells (HEK-SERT). They presented SERT with varying doses of either citalopram (an SSRI) or desipramine (an inhibitor of norepinephrine reuptake protein, NET). By examining the dose-response curves (using a normal medium as control), they were able to quantify that citalopram acted on SERT as an SSRI, and that desipramine had no effect on SERT. In a separate experiment, Horschitz et al. exposed HEK-SERT with citalopram on a long-term basis. They noticed that long-term exposure led to a down-regulation of binding sites. These results suggest some mechanism for long-term changes in the pre-synaptic neuron after drug therapy. Horschitz et al. found that after removing citalopram from the system, normal levels of SERT binding site expression returned.[12]
Depression has been suggested to be a result of a decrease of serotonin found in the synapse. This theory has been supported by the successful reduction of depressive symptoms after administration of tri-cyclic antidepressants (such as desipramine) and SSRI’s. Tri-cyclic antidepressants inhibit the reuptake of both serotonin and norepinephrine by acting upon both the SERT and NET. SSRIs selectively inhibit the reuptake of serotonin by acting upon SERT. The net result is an increased amount of serotonin in the synapse, thus increasing the probability that serotonin will interact with a serotonin receptor of the postsynaptic neuron. There are additional mechanisms by which serotonin autoreceptor desensitization must occur, but the net result is the same.[13] This increases serotonin signaling, which then acts to elevate mood and thus relieve depressive symptoms. This proposal for the antidepressant mechanism of serotonin reuptake inhibitors does not account for the time course of the therapeutic effect, which takes weeks to months, while transporter inhibition is essentially immediate.
The net effect of amphetamine (AMPH) use is an increase of dopamine, norepinephrine and serotonin in the synapse. It has been shown that AMPH acts upon trace amine-associated receptor 1 (TAAR1) to induce efflux and reuptake inhibition in the serotonin, norepinephrine, and dopamine transporters. This effect requires the transporter and TAAR1 to be co-localized (occur together) within the same neuron.
Neuroprotective role
Astrocytes seem to utilize reuptake mechanisms for a neuroprotective role. Astrocytes use GLT-1 to remove glutamate from the synapse. GLT-1 knockout mice were more prone to lethal and spontaneous seizures and acute brain injuries among the cortex. These effects could be linked to increased concentrations of glutamate in the brains of GLT-1 knockout mice, analyzed post-mortem.[14]
References
- ↑ Masson, J; Sagné, C; Hamon, M; El Mestikawy, S (1999). "Neurotransmitter transporters in the central nervous system". Pharmacological reviews. 51 (3): 439–64. PMID 10471414.
- ↑ Androutsellis-Theotokis A.; Rudnick G. (2002). "Accessibility and conformational coupling in serotonin transporter predicted internal domains". J Neurosci. 22: 8370–8378.
- ↑ Yamashita, Atsuko; Singh, Satinder K.; Kawate, Toshimitsu; Jin, Yan; Gouaux, Eric (2005). "Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters". Nature. 437 (7056): 215–23. doi:10.1038/nature03978. PMID 16041361.
- ↑ Claxton DP, Quick M, Shi L, de Carvalho FD, Weinstein H, Javitch JA, McHaourab HS (2010). "Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters". Nat Struct Mol Biol. 17 (7): 822–9. doi:10.1038/nsmb.1854.
- ↑ Zomot, E; Bendahan, A; Quick, M; Zhao, Y; Javitch, JA; Kanner, BI (2007). "Mechanism of chloride interaction with neurotransmitter:sodium symporters". Nature. 449 (7163): 726–30. doi:10.1038/nature06133. PMID 17704762.
- ↑ Tavoulari S, Rizwan AN, Forrest LR, Rudnick G (2011). "Reconstructing a Chloride-binding Site in a Bacterial Neurotransmitter Transporter Homologue". J Biol Chem. 286 (4): 2834–42. doi:10.1074/jbc.m110.186064.
- ↑ Forrest LR, Rudnick G (2009). "The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters". Physiology (Bethesda). 24: 377–86. doi:10.1152/physiol.00030.2009.
- ↑ Krishnamurthy H, Gouaux E (2012). "X-ray structures of LeuT in substrate-free outward-open and apo inward-open states". Nature. 481 (7382): 469–74. doi:10.1038/nature10737.
- ↑ Singh SK, Yamashita A, Gouaux E (2007). "Antidepressant binding site in a bacterial homologue of neurotransmitter transporters". Nature. 448 (7156): 952–6. doi:10.1038/nature06038.
- ↑ Zhou, Z; Zhen, J; Karpowich, NK; Goetz, RM; Law, CJ; Reith, ME; Wang, DN (2007). "LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake". Science. 317 (5843): 1390–3. doi:10.1126/science.1147614. PMC 3711652
 . PMID 17690258.
. PMID 17690258. - ↑ Andersen J, Kristensen AS, Bang-Andersen B, Stromgaard K. "Recent advances in the understanding of the interaction of antidepressant drugs with serotonin and norepinephrine transporters". Chem Commun. 2009 (25): 3677–92. doi:10.1039/b903035m.
- 1 2 Horschitz, S; Hummerich, R; Schloss, P (2001). "Structure, function and regulation of the 5-hydroxytryptamine (serotonin) transporter" (PDF). Biochemical Society Transactions. 29 (Pt 6): 728–32. doi:10.1042/BST0290728. PMID 11709064.
- ↑ Członkowska, AI; Zienowicz, M; Bidziński, A; MacIejak, P; Lehner, M; Taracha, E; Wisłowska, A; Płaźnik, A (2003). "The role of neurosteroids in the anxiolytic,antidepressive- and anticonvulsive effects of selective serotonin reuptake inhibitors". Medical science monitor : international medical journal of experimental and clinical research. 9 (11): RA270–5. PMID 14586292.
- ↑ Tanaka, K; Watase, K; Manabe, T; Yamada, K; Watanabe, M; Takahashi, K; Iwama, H; Nishikawa, T; et al. (1997). "Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1". Science. 276 (5319): 1699–702. doi:10.1126/science.276.5319.1699. PMID 9180080.