Synthetic lethality
Synthetic lethality arises when a combination of deficiencies in the expression of two or more genes leads to cell death, whereas a deficiency in only one of these genes does not. The deficiencies can arise through mutations, epigenetic alterations or inhibitors of one of the genes. In a synthetic lethal genetic screen, it is necessary to begin with a mutation that does not kill the cell, although may confer a phenotype (for example, slow growth), and then systematically test other mutations at additional loci to determine which confer lethality. Synthetic lethality indicates functional relationships between genes.
Background
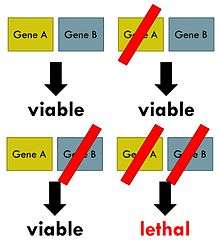
The phenomenon of synthetic lethality was first described by Calvin Bridges in 1922, who noticed that some combinations of mutations in the model organism Drosophila melanogaster confer lethality.[1] Theodore Dobzhansky coined the term "synthetic lethality" in 1946 to describe the same type of genetic interaction in wildtype populations of Drosophila.[2] If the combination of genetic events results in a non-lethal reduction in fitness, the interaction is called synthetic sickness. Although in classical genetics the term synthetic lethality refers to the interaction between two genetic perturbations, synthetic lethality can also apply to cases in which the combination of a mutation and the action of a chemical compound causes lethality, whereas the mutation or compound alone are non-lethal.[3]
Synthetic lethality is a consequence of the tendency of organisms to maintain buffering schemes that allow phenotypic stability despite genetic variation, environmental changes and random events such as mutations. This genetic robustness is the result of parallel redundant pathways and "capacitor" proteins that camouflage the effects of mutations so that important cellular processes do not depend on any individual component.[4] Synthetic lethality can help identify these buffering relationships, and what type of disease or malfunction that may occur when these relationships break down, through the identification of gene interactions that function in either the same biochemical process or pathways that appear to be unrelated.[5]
High-throughput screens
Synthetic lethality may be explored in a variety of model organisms, including Drosophila melanogaster and Saccharomyces cerevisiae. Since synthetic lethal mutations are inherently inviable, common approaches are to employ temperature sensitive mutations or put mutations under the control of a regulated promoter to allow exploration of the phenotype without leading to death.[6] Some synthetic lethal pairs are detected while attempting to elucidate molecular mechanisms of fundamental biological processes without the use of high-throughput screens.[7] For instance, the synthetic lethality of Parkin and MTF1 in Drosophila was discovered when examining the relationship between oxidative stress and metal homeostasis in the pathogenesis of Parkinson's disease.
However, high-throughput synthetic lethal screens may help illuminate questions about how cellular processes work without previous knowledge of gene function or interaction. Screening strategy must take into account the organism used for screening, the mode of genetic perturbation, and whether the screen is forward or reverse. Many of the first synthetic lethal screens were performed in S. cerevisiae. Budding yeast has many experimental advantages in screens, including a small genome, fast doubling time, both haploid and diploid states, and ease of genetic manipulation.[8] Gene ablation can be performed using a PCR-based strategy and complete libraries of knockout collections for all annotated yeast genes are publicly available. Synthetic genetic array (SGA), synthetic lethality by microarray (SLAM), and genetic interaction mapping (GIM) are three high-throughput methods for analyzing synthetic lethality in yeast. A genome scale genetic interaction map was created by SGA analysis in S. cerevisiae that comprises about 75% of all yeast genes.[9] By examining 5.4 million gene-gene pairs for synthetic lethality, an unbiased network of functional connections between genetic interactions was constructed.
High-throughput synthetic lethality screens are also performed in metazoans, but a major challenge is efficient gene perturbation. In the nematode C. elegans, RNA-interference can be combined with a query strain loss-of-function mutation. While RNA-interference is more experimentally demanding in Drosophila, living cell microarrays allow knockdown of two genes simultaneously.[10] RNA-interference is also feasible in mammalian cells, and chemical screens in mammalian cell lines is important for identifying pharmacological targets of drugs.
In chemotherapeutics
A synthetic lethal approach to cancer therapy is currently being explored as a means of developing therapies that reduce off-target effects of chemotherapies and chemopreventative drugs. Cancer cells are marked by genetic instability, errors in DNA repair, and uncontrolled transcription, which create new synthetic lethal partners in cancer cells. Because a drug effect targeting a specific gene product resembles the phenotype caused by a mutation in that gene, a cancer-related mutation can sensitize cancer cells to chemotherapeutics that target its synthetic lethal partner. Consequently, drugs that target synthetic lethal partners of mutations in cancer cells may not be toxic to normal cells, which could avoid off-target side effects of chemotherapeutics.
Synthetic lethal analysis can be used to elucidate mechanisms of known chemotherapeutic drugs by identifying genes whose function is necessary for drug function. For example, BRCA1 and BRCA2 are important for repairing double-strand breaks in DNA, and mutations in these genes predispose individuals to breast cancer and ovarian cancer.[11] The enzyme PARP1 is involved in repairing single-strand breaks, and the inhibition of PARP1 in a BRCA mutant background is selectively lethal to tumors because cancer cells accumulate DNA lesions that they cannot repair. Synthetic lethality is also useful for screening libraries of molecules to detect drugs that selectively inhibit cancer cells. In a recent chemical-genetic screen, one compound of 3200 screened molecules was a synthetic lethal inhibitor of pancreatic cancer KRAS gain-of-function cells, which suggests a potential treatment for this cancer type.[12]
Source of carcinogenic driver mutations and driver epimutations
As pointed out by Gao et al.,[13] the stability and integrity of the human genome are maintained by the DNA damage repair (DDR) system. In the presence of a defect in DDR, DNA damages will accumulate. Un-repaired DNA damage is a major cause of mutations that drive carcinogenesis.[14][15] Such excess DNA damage can increase mutational errors during DNA replication due to error-prone translesion synthesis. Excess DNA damage can also increase epigenetic alterations due to errors during DNA repair.[16][17] Such mutations and epigenetic alterations are the source of the mut-drivers and epi-drivers[18] that cause progression to cancer.
Prominence of DDR deficiencies in cancers
About 3 or 4 driver mutations and 60 passenger mutations occur in the exome (protein coding region) of a cancer.[19] However, a much larger number of mutations occur in the non-protein-coding regions of DNA in a cancer. The average number of DNA sequence mutations in the entire genome of a breast cancer tissue sample is much higher, about 20,000.[20] In an average melanoma tissue sample the total number of DNA sequence mutations is about 80,000.[21]
Early epigenetic or mutational alterations in DDR genes are likely the source of the genetic instability characteristic of cancers.[22] While a mutation or epimutation in a DDR gene, itself, would not confer a selective advantage, such a repair defect may be carried along as a passenger in a cell when the cell acquires an additional mutation/epimutation that does provide a proliferative advantage. Such cells, with both proliferative advantages and one or more DNA repair defects (causing a very high mutation rate), likely give rise to the 20,000 to 80,000 total genome mutations frequently seen in cancers. Thus a defect in a DDR gene is likely to be present in cancers (see, e.g. frequencies of epimutations in DNA repair genes in cancers). Synthetic lethality with an identified DNA repair defect in a cancer could thus be a frequent effective method for therapeutic attack on the cancer.
DDR deficiencies and synthetic lethality
DNA mismatch repair deficiency
Mutations in genes employed in DNA mismatch repair (MMR) cause a high mutation rate.[23][24] In tumors, such frequent subsequent mutations often generate "non-self" immunogenic antigens. A human Phase II clinical trial, with 41 patients, evaluated one synthetic lethal approach for tumors with or without MMR defects.[25] In the case of sporadic tumors evaluated, the majority would be deficient in MMR due to epigenetic repression of an MMR gene (see DNA mismatch repair). The product of gene PD-1 ordinarily represses cytotoxic immune responses. Inhibition of this gene allows a greater immune response. In this Phase II clinical trial with 47 patients, when cancer patients with a defect in MMR in their tumors were exposed to an inhibitor of PD-1, 67% - 78% of patients experienced immune-related progression-free survival. In contrast, for patients without defective MMR, addition of PD-1 inhibitor generated only 11% of patients with immune-related progression-free survival. Thus inhibition of PD-1 is primarily synthetically lethal with MMR defects.
Werner syndrome gene deficiency
The analysis of 630 human primary tumors in 11 tissues shows that WRN promoter hypermethylation (with loss of expression of WRN protein) is a common event in tumorigenesis.[26] The WRN gene promoter is hypermethylated in about 38% of colorectal cancers and non-small-cell lung carcinomas and in about 20% or so of stomach cancers, prostate cancers, breast cancers, non-Hodgkin lymphomas and chondrosarcomas, plus at significant levels in the other cancers evaluated. The WRN helicase protein is important in homologous recombinational DNA repair and also has roles in non-homologous end joining DNA repair and base excision DNA repair.[27]
Topoisomerase inhibitors are frequently used as chemotherapy for different cancers, though they cause bone marrow suppression, are cardiotoxic and have variable effectiveness.[28] A 2006 retrospective study, with long clinical follow-up, was made of colon cancer patients treated with the topoisomerase inhibitor irinotecan. In this study, 45 patients had hypermethylated WRN gene promoters and 43 patients had unmethylated WRN gene promoters.[26] Irinitecan was more strongly beneficial for patients with hypermethylated WRN promoters (39.4 months survival) than for those with unmethylated WRN promoters (20.7 months survival). Thus, a topoisomerase inhibitor appeared to be synthetically lethal with deficient expression of WRN. Further evaluations have also indicated synthetic lethality of deficient expression of WRN and topoisomerase inhibitors.[29][30][31][32][33]
Clinical and preclinical PARP1 inhibitor synthetic lethality
As reviewed by Murata et al.,[34] five different PARP1 inhibitors are now undergoing Phase I, II and III clinical trials, to determine if particular PARP1 inhibitors are synthetically lethal in a large variety of cancers, including those in the prostate, pancreas, non-small-cell lung tumors, lymphoma, multiple myeloma, and Ewing sarcoma. In addition, in preclinical studies using cells in culture or within mice, PARP1 inhibitors are being tested for synthetic lethality against epigenetic and mutational deficiencies in about 20 DNA repair defects beyond BRCA1/2 deficiencies. These include deficiencies in PALB2, FANCD2, RAD51, ATM, MRE11, p53, XRCC1 and LSD1.
Preclinical ARID1A synthetic lethality
ARID1A, a chromatin modifier, is required for non-homologous end joining, a major pathway that repairs double-strand breaks in DNA,[35] and also has transcription regulatory roles.[36] ARID1A mutations are one of the 12 most common carcinogenic mutations.[37] Mutation or epigenetically decreased expression[38] of ARID1A has been found in 17 types of cancer.[39] Pre-clinical studies in cells and in mice show that synthetic lethality for deficient ARID1A expression occurs by either inhibition of the methyltransferase activity of EZH2,[40][41] or with addition of the kinase inhibitor dasatinib.[42]
Preclinical RAD52 synthetic lethality
There are two pathways for homologous recombinational repair of double-strand breaks. The major pathway depends on BRCA1, PALB2 and BRCA2 while an alternative pathway depends on RAD52.[43] Pre-clinical studies, involving epigenetically reduced or mutated BRCA-deficient cells (in culture or injected into mice), show that inhibition of RAD52 is synthetically lethal with BRCA-deficiency.[44]
Synthetic lethality side effects
Although treatments using synthetic lethality can stop or slow progression of cancers and prolong survival, each of the synthetic lethal treatments has some adverse side effects. For example, more than 20% of patients treated with an inhibitor of PD-1 encounter fatigue, rash, pruritus, cough, diarrhea, decreased appetite, constipation or arthralgia .[45] Thus, it is important to determine which DDR deficiency is present, so that only an effective synthetic lethal treatment can be applied, and not unnecessarily subject patients to adverse side effects without a direct benefit.
See also
References
- ↑ Nijman, Sebastian (Jan 3, 2011). "Synthetic Lethality: General principles, utility and detection using genetic screens in human cells". FEBS Lett. 585 (1): 1–6. doi:10.1016/j.febslet.2010.11.024. PMID 21094158.
- ↑ Ferrari, Elisa; Lucca, Chiara; Foiani, Marco (Nov 2010). "A lethal combination for cancer cells: synthetic lethality screenings for drug discovery". Eur J Cancer. 46 (16): 2889–95. doi:10.1016/j.ejca.2010.07.031. PMID 20724143.
- ↑ Hartwell, LH (Nov 7, 1997). "Integrating genetic approaches into the discovery of anticancer drugs" (PDF). Science. 278 (5340). doi:10.1126/science.278.5340.1064. Retrieved 10 September 2014.
- ↑ Baugh, LR (2005). "Synthetic lethal analysis of Caenorhabditis elegans posterior embryonic patterning genes identifies conserved genetic interactions". Genome Biol. 6 (5): R45. doi:10.1186/gb-2005-6-5-r45. PMID 15892873. Retrieved 10 September 2014.
- ↑ Hartman; Garvik, B; Hartwell, L (Feb 2001). "Principles for the buffering of genetic variation". Science. 291 (5506): 1001–4. doi:10.1126/science.291.5506.1001.
- ↑ http://www.sci.sdsu.edu/~smaloy/MicrobialGenetics/topics/rev-sup/synthetic.html "Synthetic Lethal Mutations." Retrieved on 2010-01-27.
- ↑ Saini, N; Georgiev, O; Schaffner, W (May 2011). "The parkin mutant phenotype in the fly is largely rescued by metal-responsive transcription factor (MTF-1)". Mol Cell Biol. 31 (10): 2151–61. doi:10.1128/MCB.05207-11. PMID 21383066.
- ↑ Matuo, Renata; Sousa, Fabricio; Soares, Daniele; Bonatto, Diego; Saffi, Jenifer; Escargueil, Alexandre; Larsen, Annette; Henriques, Joao (Oct 2012). "Saccharomyces cerevisiae as a model system to study the response to anticancer agents". Cancer Chemother Pharmacol. 70 (4): 491–502. doi:10.1007/s00280-012-1937-4. PMID 22851206. Retrieved 15 November 2014.
- ↑ Costanzo, Michael (Jan 2010). "The Genetic Landscape of a Cell". Science. 327 (5964): 425–431. doi:10.1126/science.1180823. PMID 20093466. Retrieved 16 November 2014.
- ↑ Wheeler, Douglas; Bailey, Steve; Guertin, David; Carpenter, Anne; OHiggins, Caitlin; Sabatini, David (Oct 2004). "RNAi living-cell microarrays for loss-of-function screens in Drosophila melanogaster cells". Nat Methods. 1 (2): 127–132. doi:10.1038/nmeth711. PMID 15782175. Retrieved 16 November 2014.
- ↑ Farmer, Hannah; McCabe, Nuala; Lord, Christopher; Tutt, Andrew; Johnson, Damian; Richardson, Tobias; Santarosa, Manuela; Dillon, Krystyna; Hickson, Ian; Knights, Charlotte; Martin, Niall; Jackson, Stephen; Smith, Graeme; Ashworth, Alan (Apr 2005). "Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy". Nature. 434 (7035): 917–921. doi:10.1038/nature03445. PMID 15829967. Retrieved 16 November 2014.
- ↑ Ji, Zhenyu; Mei, Fang; Lory, Pedro; Gilbertson, Scott; Chen, Yijun; Cheng, Xiaodong (Jan 2009). "Chemical genetic screening of KRAS-based synthetic lethal inhibitors for pancreatic cancer". Front Biosci (Landmark Ed). 14: 2904–10. PMID 19273243. Retrieved 16 November 2014.
- ↑ Gao D, Herman JG, Guo M (2016). "The clinical value of aberrant epigenetic changes of DNA damage repair genes in human cancer". Oncotarget. doi:10.18632/oncotarget.7949. PMID 26967246.
- ↑ Kastan MB (2008). "DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture". Mol. Cancer Res. 6 (4): 517–24. doi:10.1158/1541-7786.MCR-08-0020. PMID 18403632.
- ↑ Bernstein, C; Prasad, AR; Nfonsam, V; Bernstein, H. (2013). "Chapter 16: DNA Damage, DNA Repair and Cancer". In Chen, Clark. New Research Directions in DNA Repair,. p. 413. ISBN 978-953-51-1114-6.
- ↑ O'Hagan HM, Mohammad HP, Baylin SB (2008). "Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island". PLoS Genetics. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723
 . PMID 18704159.
. PMID 18704159. - ↑ Cuozzo C, Porcellini A, Angrisano T, et al. (July 2007). "DNA damage, homology-directed repair, and DNA methylation". PLoS Genetics. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- ↑ Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (2013). "Cancer genome landscapes". Science. 339 (6127): 1546–58. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
- ↑ Vogelstein B; Papadopoulos N; Velculescu VE; Zhou S; Diaz LA; Kinzler KW (March 2013). "Cancer genome landscapes". Science. 339 (6127): 1546–58. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
- ↑ Yost SE; Smith EN; Schwab RB; Bao L; Jung H; Wang X; Voest E; Pierce JP; Messer K; Parker BA; Harismendy O; Frazer KA (August 2012). "Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens". Nucleic Acids Res. 40 (14): e107. doi:10.1093/nar/gks299. PMC 3413110. PMID 22492626.
- ↑ Berger MF; Hodis E; Heffernan TP; Deribe YL; Lawrence MS; Protopopov A; Ivanova E; Watson IR; Nickerson E; Ghosh P; Zhang H; Zeid R; Ren X; Cibulskis K; Sivachenko AY; Wagle N; Sucker A; Sougnez C; Onofrio R; Ambrogio L; Auclair D; Fennell T; Carter SL; Drier Y; Stojanov P; Singer MA; Voet D; Jing R; Saksena G; Barretina J; Ramos AH; Pugh TJ; Stransky N; Parkin M; Winckler W; Mahan S; Ardlie K; Baldwin J; Wargo J; Schadendorf D; Meyerson M; Gabriel SB; Golub TR; Wagner SN; Lander ES; Getz G; Chin L; Garraway LA (May 2012). "Melanoma genome sequencing reveals frequent PREX2 mutations". Nature. 485 (7399): 502–6. doi:10.1038/nature11071. PMC 3367798. PMID 22622578.
- ↑ Bernstein C, Prasad AR, Nfonsam V, Bernstein H. (2013). DNA Damage, DNA Repair and Cancer, New Research Directions in DNA Repair, Prof. Clark Chen (Ed.), ISBN 978-953-51-1114-6, InTech, http://www.intechopen.com/books/new-research-directions-in-dna-repair/dna-damage-dna-repair-and-cancer
- ↑ Narayanan, L.; Fritzell, J. A.; Baker, S. M.; Liskay, R. M.; Glazer, P. M. (1997). "Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2". Proceedings of the National Academy of Sciences. 94 (7): 3122–3127. doi:10.1073/pnas.94.7.3122. PMC 20332. PMID 9096356.
- ↑ Hegan, D. C.; Narayanan, L.; Jirik, F. R.; Edelmann, W.; Liskay, R.M.; Glazer, P. M. (2006). "Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6". Carcinogenesis. 27 (12): 2402–2408. doi:10.1093/carcin/bgl079. PMC 2612936. PMID 16728433.
- ↑ Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA (2015). "PD-1 Blockade in Tumors with Mismatch-Repair Deficiency". N. Engl. J. Med. 372 (26): 2509–20. doi:10.1056/NEJMoa1500596. PMC 4481136. PMID 26028255.
- 1 2 Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF, Herranz M, Paz MF, Sanchez-Cespedes M, Artiga MJ, Guerrero D, Castells A, von Kobbe C, Bohr VA, Esteller M (2006). "Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer". Proc. Natl. Acad. Sci. U.S.A. 103 (23): 8822–7. doi:10.1073/pnas.0600645103. PMC 1466544. PMID 16723399.
- ↑ Monnat RJ (2010). "Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology". Semin. Cancer Biol. 20 (5): 329–39. doi:10.1016/j.semcancer.2010.10.002. PMC 3040982. PMID 20934517.
- ↑ Pommier Y (2013). "Drugging topoisomerases: lessons and challenges". ACS Chem. Biol. 8 (1): 82–95. doi:10.1021/cb300648v. PMC 3549721. PMID 23259582.
- ↑ Wang L, Xie L, Wang J, Shen J, Liu B (2013). "Correlation between the methylation of SULF2 and WRN promoter and the irinotecan chemosensitivity in gastric cancer". BMC Gastroenterol. 13: 173. doi:10.1186/1471-230X-13-173. PMC 3877991. PMID 24359226.
- ↑ Bird JL, Jennert-Burston KC, Bachler MA, Mason PA, Lowe JE, Heo SJ, Campisi J, Faragher RG, Cox LS (2012). "Recapitulation of Werner syndrome sensitivity to camptothecin by limited knockdown of the WRN helicase/exonuclease". Biogerontology. 13 (1): 49–62. doi:10.1007/s10522-011-9341-8. PMID 21786128.
- ↑ Masuda K, Banno K, Yanokura M, Tsuji K, Kobayashi Y, Kisu I, Ueki A, Yamagami W, Nomura H, Tominaga E, Susumu N, Aoki D (2012). "Association of epigenetic inactivation of the WRN gene with anticancer drug sensitivity in cervical cancer cells". Oncol. Rep. 28 (4): 1146–52. doi:10.3892/or.2012.1912. PMC 3583574. PMID 22797812.
- ↑ Futami K, Takagi M, Shimamoto A, Sugimoto M, Furuichi Y (2007). "Increased chemotherapeutic activity of camptothecin in cancer cells by siRNA-induced silencing of WRN helicase". Biol. Pharm. Bull. 30 (10): 1958–61. PMID 17917271.
- ↑ Futami K, Ishikawa Y, Goto M, Furuichi Y, Sugimoto M (2008). "Role of Werner syndrome gene product helicase in carcinogenesis and in resistance to genotoxins by cancer cells". Cancer Sci. 99 (5): 843–8. doi:10.1111/j.1349-7006.2008.00778.x. PMID 18312465.
- ↑ Murata S, Zhang C, Finch N, Zhang K, Campo L, Breuer EK (2016). "Predictors and Modulators of Synthetic Lethality: An Update on PARP Inhibitors and Personalized Medicine". Biomed Res Int. 2016: 2346585. doi:10.1155/2016/2346585. PMC 5013223. PMID 27642590.
- ↑ Watanabe R, Ui A, Kanno S, Ogiwara H, Nagase T, Kohno T, Yasui A (2014). "SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability". Cancer Res. 74 (9): 2465–75. doi:10.1158/0008-5472.CAN-13-3608. PMID 24788099.
- ↑ Raab JR, Resnick S, Magnuson T (2015). "Genome-Wide Transcriptional Regulation Mediated by Biochemically Distinct SWI/SNF Complexes". PLoS Genet. 11 (12): e1005748. doi:10.1371/journal.pgen.1005748. PMC 4699898. PMID 26716708.
- ↑ Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G (2014). "Discovery and saturation analysis of cancer genes across 21 tumour types". Nature. 505 (7484): 495–501. doi:10.1038/nature12912. PMC 4048962. PMID 24390350.
- ↑ Zhang X, Sun Q, Shan M, Niu M, Liu T, Xia B, Liang X, Wei W, Sun S, Zhang Y, Liu XS, Song Q, Yang Y, Ma Y, Liu Y, Yang L, Ren Y, Zhang G, Pang D (2013). "Promoter hypermethylation of ARID1A gene is responsible for its low mRNA expression in many invasive breast cancers". PLoS ONE. 8 (1): e53931. doi:10.1371/journal.pone.0053931. PMC 3549982. PMID 23349767.
- ↑ Wu JN, Roberts CW (2013). "ARID1A mutations in cancer: another epigenetic tumor suppressor?". Cancer Discov. 3 (1): 35–43. doi:10.1158/2159-8290.CD-12-0361. PMC 3546152. PMID 23208470.
- ↑ Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih IeM, Conejo-Garcia JR, Speicher DW, Zhang R (2015). "Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers". Nat. Med. 21 (3): 231–8. doi:10.1038/nm.3799. PMC 4352133. PMID 25686104.
- ↑ Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, Wang W, Haswell JR, Walensky LD, Hahn WC, Orkin SH, Roberts CW (2015). "SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2". Nat. Med. 21 (12): 1491–6. doi:10.1038/nm.3968. PMC 4886303. PMID 26552009.
- ↑ Miller RE, Brough R, Bajrami I, Williamson CT, McDade S, Campbell J, Kigozi A, Rafiq R, Pemberton H, Natrajan R, Joel J, Astley H, Mahoney C, Moore JD, Torrance C, Gordan JD, Webber JT, Levin RS, Shokat KM, Bandyopadhyay S, Lord CJ, Ashworth A (2016). "Synthetic Lethal Targeting of ARID1A-Mutant Ovarian Clear Cell Tumors with Dasatinib". Mol. Cancer Ther. 15 (7): 1472–84. doi:10.1158/1535-7163.MCT-15-0554. PMID 27364904.
- ↑ Lok BH, Carley AC, Tchang B, Powell SN (2013). "RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination". Oncogene. 32 (30): 3552–8. doi:10.1038/onc.2012.391. PMID 22964643.
- ↑ Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, Padget M, Irvine DA, Sliwinski T, Haas K, Lee J, Geng H, Roy D, Slupianek A, Rassool FV, Wasik MA, Childers W, Copland M, Müschen M, Civin CI, Skorski T (2013). "Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile". Blood. 122 (7): 1293–304. doi:10.1182/blood-2013-05-501072. PMC 3744994. PMID 23836560.
- ↑ Villadolid J, Amin A (2015). "Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities". Transl Lung Cancer Res. 4 (5): 560–75. doi:10.3978/j.issn.2218-6751.2015.06.06. PMC 4630514. PMID 26629425.
External links
- Data Repository of Yeast Genetic INteractions
- Saccharomyces Genome Deletion Project
- Syn-Lethality Database
| Look up synthetic lethality in Wiktionary, the free dictionary. |