Polymerase chain reaction

Polymerase chain reaction (PCR) is a technique used in molecular biology to amplify a single copy or a few copies of a piece of DNA across several orders of magnitude, generating thousands to millions of copies of a particular DNA sequence. It is an easy and cheap tool to amplify a focused segment of DNA, useful for such purposes as the diagnosis and monitoring of genetic diseases, identification of criminals (in the field of forensics), and studying the function of a targeted segment of DNA.[1]
Developed in 1983 by Kary Mullis,[2][3] PCR is now a common and often indispensable technique used in clinical laboratories and research laboratories for a variety of applications.[4][5] These include DNA cloning for sequencing, DNA-based phylogeny, or functional analysis of genes; the diagnosis of hereditary diseases; the identification of genetic fingerprints (used in forensic sciences and DNA paternity testing); and the detection of pathogens in nucleic acid tests for the diagnosis of infectious diseases. In 1993, Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR.[6]
The method relies on thermal cycling, consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA. Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase, which the method is named after, are key components to enable selective and repeated amplification. As PCR progresses, the DNA generated is itself used as a template for replication, setting in motion a chain reaction in which the DNA template is exponentially amplified. PCR can be extensively modified to perform a wide array of genetic manipulations. PCR is not generally considered to be a recombinant DNA method, as it does not involve cutting and pasting DNA, only amplification of existing sequences.
Almost all PCR applications employ a heat-stable DNA polymerase, such as Taq polymerase (an enzyme originally isolated from the bacterium Thermus aquaticus). This DNA polymerase enzymatically assembles a new DNA strand from DNA building-blocks, the nucleotides, by using single-stranded DNA as a template and DNA oligonucleotides (also called DNA primers), which are required for initiation of DNA synthesis. The vast majority of PCR methods use thermal cycling, i.e., alternately heating and cooling the PCR sample through a defined series of temperature steps.
In the first step, the two strands of the DNA double helix are physically separated at a high temperature in a process called DNA melting. In the second step, the temperature is lowered and the two DNA strands become templates for DNA polymerase to selectively amplify the target DNA. The selectivity of PCR results from the use of primers that are complementary to the DNA region targeted for amplification under specific thermal cycling conditions.

Principles


PCR amplifies a specific region of a DNA strand (the DNA target). Most PCR methods typically amplify DNA fragments of between 0.1 and 10 kilo base pairs (kbp), although some techniques allow for amplification of fragments up to 40 kbp in size.[7] The amount of amplified product is determined by the available substrates in the reaction, which become limiting as the reaction progresses.[8]
A basic PCR set up requires several components and reagents.[9] These components include:
- DNA template that contains the DNA region (target) to amplify
- Two primers that are complementary to the 3' (three prime) ends of each of the sense and anti-sense strand of the DNA target. Primers can be custom made in a laboratory that are complementary to the DNA segment to be amplified.
- Taq polymerase ([1] as the DNA Polymerase can not attach to a DNA strand and elongate on its own. It should also be heat resistant,[10] so that it can withstand the denaturation process.
- Deoxynucleoside triphosphates (dNTPs, sometimes called "deoxynucleotide triphosphates"; nucleotides containing triphosphate groups), the building-blocks from which the DNA polymerase synthesizes a new DNA strand.
- Buffer solution, providing a suitable chemical environment for optimum activity and stability of the DNA polymerase
- Bivalent cations, magnesium or manganese ions; generally Mg2+ is used, but Mn2+ can be used for PCR-mediated DNA mutagenesis, as higher Mn2+ concentration increases the error rate during DNA synthesis[11]
- Monovalent cation potassium ions
The PCR is commonly carried out in a reaction volume of 10–200 μl in small reaction tubes (0.2–0.5 ml volumes) in a thermal cycler. The thermal cycler heats and cools the reaction tubes to achieve the temperatures required at each step of the reaction (see below). Many modern thermal cyclers make use of the Peltier effect, which permits both heating and cooling of the block holding the PCR tubes simply by reversing the electric current. Thin-walled reaction tubes permit favorable thermal conductivity to allow for rapid thermal equilibration. Most thermal cyclers have heated lids to prevent condensation at the top of the reaction tube. Older thermocyclers lacking a heated lid require a layer of oil on top of the reaction mixture or a ball of wax inside the tube.
Procedure
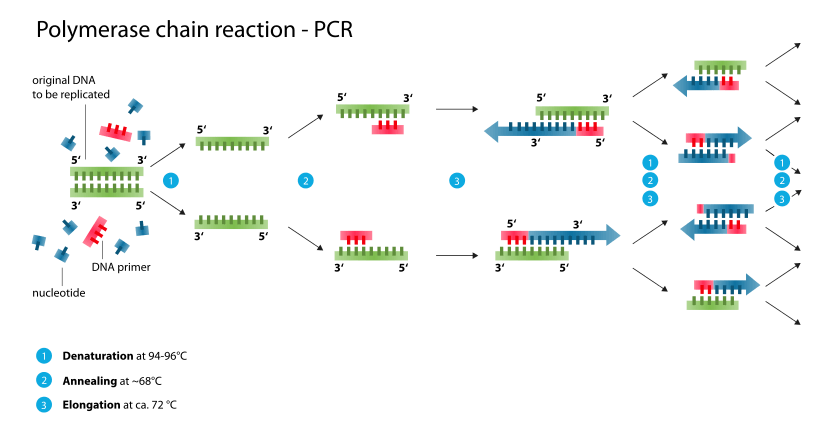
Typically, PCR consists of a series of 20–40 repeated temperature changes, called cycles, with each cycle commonly consisting of 2–3 discrete temperature steps, usually three (Figure below). The cycling is often preceded by a single temperature step at a high temperature (>90 °C), and followed by one hold at the end for final product extension or brief storage. The temperatures used and the length of time they are applied in each cycle depend on a variety of parameters. These include the enzyme used for DNA synthesis, the concentration of divalent ions and dNTPs in the reaction, and the melting temperature (Tm) of the primers.[12]
- Initialization step (Only required for DNA polymerases that require heat activation by hot-start PCR.[13]): This step consists of heating the reaction to a temperature of 94–96 °C (or 98 °C if extremely thermostable polymerases are used), which is held for 1–9 minutes.
- Denaturation step: This step is the first regular cycling event and consists of heating the reaction to 94–98 °C for 20–30 seconds. It causes DNA melting of the DNA template by disrupting the hydrogen bonds between complementary bases, yielding single-stranded DNA molecules.
- Annealing step: The reaction temperature is lowered to 50–65 °C for 20–40 seconds allowing annealing of the primers to the single-stranded DNA template. This temperature must be low enough to allow for hybridization of the primer to the strand, but high enough for the hybridization to be specific, i.e., the primer should only bind to a perfectly complementary part of the template. If the temperature is too low, the primer could bind imperfectly. If it is too high, the primer might not bind. Typically the annealing temperature is about 3–5 °C below the Tm of the primers used. Stable DNA–DNA hydrogen bonds are only formed when the primer sequence very closely matches the template sequence. The polymerase binds to the primer-template hybrid and begins DNA formation. It is very vital to determine the annealing temperature in PCR. This is because in PCR, efficiency and specificity are affected by the annealing temperature. An incorrect annealing temperature will cause an error in the test.
- Extension/elongation step: The temperature at this step depends on the DNA polymerase used; Taq polymerase has its optimum activity temperature at 75–80 °C,[14][15] and commonly a temperature of 72 °C is used with this enzyme. At this step the DNA polymerase synthesizes a new DNA strand complementary to the DNA template strand by adding dNTPs that are complementary to the template in 5' to 3' direction, condensing the 5'-phosphate group of the dNTPs with the 3'-hydroxyl group at the end of the nascent (extending) DNA strand. The extension time depends both on the DNA polymerase used and on the length of the DNA fragment to amplify. As a rule-of-thumb, at its optimum temperature, the DNA polymerase polymerizes a thousand bases per minute. Under optimum conditions, i.e., if there are no limitations due to limiting substrates or reagents, at each extension step, the amount of DNA target is doubled, leading to exponential (geometric) amplification of the specific DNA fragment.
The processes of denaturation, annealing and elongation constitute of one cycle. Multiple cycles are required to amplify DNA. The formula used to calculate the number of DNA copies formed after a given # of cycles repeated is, 2^# of cycles.[16]
- Final elongation: This single step is occasionally performed at a temperature of 70–74 °C (this is the temperature needed for optimal activity for most polymerases used in PCR) for 5–15 minutes after the last PCR cycle to ensure that any remaining single-stranded DNA is fully extended.
- Final hold: This step at 4–15 °C for an indefinite time may be employed for short-term storage of the reaction.
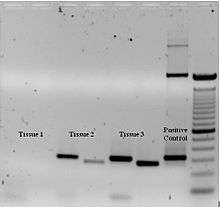
To check whether the PCR generated the anticipated DNA fragment (also sometimes referred to as the amplimer or amplicon), agarose gel electrophoresis is employed for size separation of the PCR products. The size(s) of PCR products is determined by comparison with a DNA ladder (a molecular weight marker), which contains DNA fragments of known size, run on the gel alongside the PCR products (see Fig. 3).
Stages
The PCR process can be divided into three stages:
Exponential amplification: At every cycle, the amount of product is doubled (assuming 100% reaction efficiency). The reaction is very sensitive: only minute quantities of DNA must be present.[17]
Leveling off stage: The reaction slows as the DNA polymerase loses activity and as consumption of reagents such as dNTPs and primers causes them to become limiting.
Plateau: No more product accumulates due to exhaustion of reagents and enzyme.
Optimization
In practice, PCR can fail for various reasons, in part due to its sensitivity to contamination causing amplification of spurious DNA products. Because of this, a number of techniques and procedures have been developed for optimizing PCR conditions.[18][19] Contamination with extraneous DNA is addressed with lab protocols and procedures that separate pre-PCR mixtures from potential DNA contaminants.[9] This usually involves spatial separation of PCR-setup areas from areas for analysis or purification of PCR products, use of disposable plasticware, and thoroughly cleaning the work surface between reaction setups. Primer-design techniques are important in improving PCR product yield and in avoiding the formation of spurious products, and the usage of alternate buffer components or polymerase enzymes can help with amplification of long or otherwise problematic regions of DNA. Addition of reagents, such as formamide, in buffer systems may increase the specificity and yield of PCR.[20] Computer simulations of theoretical PCR results (Electronic PCR) may be performed to assist in primer design.[21]
Applications
Selective DNA isolation
PCR allows isolation of DNA fragments from genomic DNA by selective amplification of a specific region of DNA. This use of PCR augments many methods, such as generating hybridization probes for Southern or northern hybridization and DNA cloning, which require larger amounts of DNA, representing a specific DNA region. PCR supplies these techniques with high amounts of pure DNA, enabling analysis of DNA samples even from very small amounts of starting material.
Other applications of PCR include DNA sequencing to determine unknown PCR-amplified sequences in which one of the amplification primers may be used in Sanger sequencing, isolation of a DNA sequence to expedite recombinant DNA technologies involving the insertion of a DNA sequence into a plasmid, phage, or cosmid (depending on size) or the genetic material of another organism. Bacterial colonies (such as E. coli) can be rapidly screened by PCR for correct DNA vector constructs.[22] PCR may also be used for genetic fingerprinting; a forensic technique used to identify a person or organism by comparing experimental DNAs through different PCR-based methods.
Some PCR 'fingerprints' methods have high discriminative power and can be used to identify genetic relationships between individuals, such as parent-child or between siblings, and are used in paternity testing (Fig. 4). This technique may also be used to determine evolutionary relationships among organisms when certain molecular clocks are used (i.e., the 16S rRNA and recA genes of microorganisms).[23]

Amplification and quantification of DNA
Because PCR amplifies the regions of DNA that it targets, PCR can be used to analyze extremely small amounts of sample. This is often critical for forensic analysis, when only a trace amount of DNA is available as evidence. PCR may also be used in the analysis of ancient DNA that is tens of thousands of years old. These PCR-based techniques have been successfully used on animals, such as a forty-thousand-year-old mammoth, and also on human DNA, in applications ranging from the analysis of Egyptian mummies to the identification of a Russian tsar and the body of English king Richard III.[24]
Quantitative PCR methods allow the estimation of the amount of a given sequence present in a sample—a technique often applied to quantitatively determine levels of gene expression. Quantitative PCR is an established tool for DNA quantification that measures the accumulation of DNA product after each round of PCR amplification.
Medical applications
PCR has been applied to a large number of medical procedures:
- The first application of PCR was for genetic testing, where a sample of DNA is analyzed for the presence of genetic disease mutations.[4] Prospective parents can be tested for being genetic carriers, or their children might be tested for actually being affected by a disease. DNA samples for prenatal testing can be obtained by amniocentesis, chorionic villus sampling, or even by the analysis of rare fetal cells circulating in the mother's bloodstream. PCR analysis is also essential to preimplantation genetic diagnosis, where individual cells of a developing embryo are tested for mutations.
- PCR can also be used as part of a sensitive test for tissue typing, vital to organ transplantation. As of 2008, there is even a proposal to replace the traditional antibody-based tests for blood type with PCR-based tests.[25]
- Many forms of cancer involve alterations to oncogenes. By using PCR-based tests to study these mutations, therapy regimens can sometimes be individually customized to a patient. PCR permits early diagnosis of malignant diseases such as leukemia and lymphomas, which is currently the highest-developed in cancer research and is already being used routinely. PCR assays can be performed directly on genomic DNA samples to detect translocation-specific malignant cells at a sensitivity that is at least 10,000 fold higher than that of other methods.[26]
Infectious disease applications
PCR allows for rapid and highly specific diagnosis of infectious diseases, including those caused by bacteria or viruses.[27] PCR also permits identification of non-cultivatable or slow-growing microorganisms such as mycobacteria, anaerobic bacteria, or viruses from tissue culture assays and animal models. The basis for PCR diagnostic applications in microbiology is the detection of infectious agents and the discrimination of non-pathogenic from pathogenic strains by virtue of specific genes.[27][28]
Characterization and detection of infectious disease organisms have been revolutionized by PCR in the follow ways:
- The human immunodeficiency virus (or HIV), is a difficult target to find and eradicate. The earliest tests for infection relied on the presence of antibodies to the virus circulating in the bloodstream. However, antibodies don't appear until many weeks after infection, maternal antibodies mask the infection of a newborn, and therapeutic agents to fight the infection don't affect the antibodies. PCR tests have been developed that can detect as little as one viral genome among the DNA of over 50,000 host cells.[29] Infections can be detected earlier, donated blood can be screened directly for the virus, newborns can be immediately tested for infection, and the effects of antiviral treatments can be quantified.
- Some disease organisms, such as that for tuberculosis, are difficult to sample from patients and slow to be grown in the laboratory. PCR-based tests have allowed detection of small numbers of disease organisms (both live or dead), in convenient samples. Detailed genetic analysis can also be used to detect antibiotic resistance, allowing immediate and effective therapy. The effects of therapy can also be immediately evaluated.
- The spread of a disease organism through populations of domestic or wild animals can be monitored by PCR testing. In many cases, the appearance of new virulent sub-types can be detected and monitored. The sub-types of an organism that were responsible for earlier epidemics can also be determined by PCR analysis.
- Viral DNA can be detected by PCR. The primers used must be specific to the targeted sequences in the DNA of a virus, and PCR can be used for diagnostic analyses or DNA sequencing of the viral genome. The high sensitivity of PCR permits virus detection soon after infection and even before the onset of disease.[27] Such early detection may give physicians a significant lead time in treatment. The amount of virus ("viral load") in a patient can also be quantified by PCR-based DNA quantitation techniques (see below).
Forensic applications
The development of PCR-based genetic (or DNA) fingerprinting protocols has seen widespread application in forensics:
- In its most discriminating form, genetic fingerprinting can uniquely discriminate any one person from the entire population of the world. Minute samples of DNA can be isolated from a crime scene, and compared to that from suspects, or from a DNA database of earlier evidence or convicts. Simpler versions of these tests are often used to rapidly rule out suspects during a criminal investigation. Evidence from decades-old crimes can be tested, confirming or exonerating the people originally convicted.
- Less discriminating forms of DNA fingerprinting can help in DNA paternity testing, where an individual is matched with their close relatives. DNA from unidentified human remains can be tested, and compared with that from possible parents, siblings, or children. Similar testing can be used to confirm the biological parents of an adopted (or kidnapped) child. The actual biological father of a newborn can also be confirmed (or ruled out).
Research applications
PCR has been applied to many areas of research in molecular genetics:
- PCR allows rapid production of short pieces of DNA, even when not more than the sequence of the two primers is known. This ability of PCR augments many methods, such as generating hybridization probes for Southern or northern blot hybridization. PCR supplies these techniques with large amounts of pure DNA, sometimes as a single strand, enabling analysis even from very small amounts of starting material.
- The task of DNA sequencing can also be assisted by PCR. Known segments of DNA can easily be produced from a patient with a genetic disease mutation. Modifications to the amplification technique can extract segments from a completely unknown genome, or can generate just a single strand of an area of interest.
- PCR has numerous applications to the more traditional process of DNA cloning. It can extract segments for insertion into a vector from a larger genome, which may be only available in small quantities. Using a single set of 'vector primers', it can also analyze or extract fragments that have already been inserted into vectors. Some alterations to the PCR protocol can generate mutations (general or site-directed) of an inserted fragment.
- Sequence-tagged sites is a process where PCR is used as an indicator that a particular segment of a genome is present in a particular clone. The Human Genome Project found this application vital to mapping the cosmid clones they were sequencing, and to coordinating the results from different laboratories.
- An exciting application of PCR is the phylogenic analysis of DNA from ancient sources, such as that found in the recovered bones of Neanderthals, or from frozen tissues of mammoths. In some cases the highly degraded DNA from these sources might be reassembled during the early stages of amplification.
- A common application of PCR is the study of patterns of gene expression. Tissues (or even individual cells) can be analyzed at different stages to see which genes have become active, or which have been switched off. This application can also use quantitative PCR to quantitate the actual levels of expression
- The ability of PCR to simultaneously amplify several loci from individual sperm[30] has greatly enhanced the more traditional task of genetic mapping by studying chromosomal crossovers after meiosis. Rare crossover events between very close loci have been directly observed by analyzing thousands of individual sperms. Similarly, unusual deletions, insertions, translocations, or inversions can be analyzed, all without having to wait (or pay for) the long and laborious processes of fertilization, embryogenesis, etc.
Limitations
DNA polymerase is prone to error, which in turn causes mutations in the PCR fragments that are made. Additionally, the specificity of the PCR fragments can mutate to the template DNA, due to nonspecific binding of primers. Furthermore, prior information on the sequence is necessary in order to generate the primers.[31]
Variations
- Allele-specific PCR: a diagnostic or cloning technique based on single-nucleotide variations (SNVs not to be confused with SNPs) (single-base differences in a patient). It requires prior knowledge of a DNA sequence, including differences between alleles, and uses primers whose 3' ends encompass the SNV (base pair buffer around SNV usually incorporated). PCR amplification under stringent conditions is much less efficient in the presence of a mismatch between template and primer, so successful amplification with an SNP-specific primer signals presence of the specific SNP in a sequence.[32] See SNP genotyping for more information.
- Assembly PCR or Polymerase Cycling Assembly (PCA): artificial synthesis of long DNA sequences by performing PCR on a pool of long oligonucleotides with short overlapping segments. The oligonucleotides alternate between sense and antisense directions, and the overlapping segments determine the order of the PCR fragments, thereby selectively producing the final long DNA product.[33]
- Asymmetric PCR: preferentially amplifies one DNA strand in a double-stranded DNA template. It is used in sequencing and hybridization probing where amplification of only one of the two complementary strands is required. PCR is carried out as usual, but with a great excess of the primer for the strand targeted for amplification. Because of the slow (arithmetic) amplification later in the reaction after the limiting primer has been used up, extra cycles of PCR are required.[34] A recent modification on this process, known as Linear-After-The-Exponential-PCR (LATE-PCR), uses a limiting primer with a higher melting temperature (Tm) than the excess primer to maintain reaction efficiency as the limiting primer concentration decreases mid-reaction.[35]
- Dial-out PCR: a highly parallel method for retrieving accurate DNA molecules for gene synthesis. A complex library of DNA molecules is modified with unique flanking tags before massively parallel sequencing. Tag-directed primers then enable the retrieval of molecules with desired sequences by PCR.[36]
- Digital PCR (dPCR): used to measure the quantity of a target DNA sequence in a DNA sample. The DNA sample is highly diluted so that after running many PCRs in parallel, some of them do not receive a single molecule of the target DNA. The target DNA concentration is calculated using the proportion of negative outcomes. Hence the name 'digital PCR'.
- Helicase-dependent amplification: similar to traditional PCR, but uses a constant temperature rather than cycling through denaturation and annealing/extension cycles. DNA helicase, an enzyme that unwinds DNA, is used in place of thermal denaturation.[37]
- Hot start PCR: a technique that reduces non-specific amplification during the initial set up stages of the PCR. It may be performed manually by heating the reaction components to the denaturation temperature (e.g., 95 °C) before adding the polymerase.[38] Specialized enzyme systems have been developed that inhibit the polymerase's activity at ambient temperature, either by the binding of an antibody[13][39] or by the presence of covalently bound inhibitors that dissociate only after a high-temperature activation step. Hot-start/cold-finish PCR is achieved with new hybrid polymerases that are inactive at ambient temperature and are instantly activated at elongation temperature.
- In silico PCR (digital PCR, virtual PCR, electronic PCR, e-PCR) refers to computational tools used to calculate theoretical polymerase chain reaction results using a given set of primers (probes) to amplify DNA sequences from a sequenced genome or transcriptome. In silico PCR was proposed as an educational tool for molecular biology.[40]
- Intersequence-specific PCR (ISSR): a PCR method for DNA fingerprinting that amplifies regions between simple sequence repeats to produce a unique fingerprint of amplified fragment lengths.[41]
- Inverse PCR: is commonly used to identify the flanking sequences around genomic inserts. It involves a series of DNA digestions and self ligation, resulting in known sequences at either end of the unknown sequence.[42]
- Ligation-mediated PCR: uses small DNA linkers ligated to the DNA of interest and multiple primers annealing to the DNA linkers; it has been used for DNA sequencing, genome walking, and DNA footprinting.[43]
- Methylation-specific PCR (MSP): developed by Stephen Baylin and Jim Herman at the Johns Hopkins School of Medicine,[44] and is used to detect methylation of CpG islands in genomic DNA. DNA is first treated with sodium bisulfite, which converts unmethylated cytosine bases to uracil, which is recognized by PCR primers as thymine. Two PCRs are then carried out on the modified DNA, using primer sets identical except at any CpG islands within the primer sequences. At these points, one primer set recognizes DNA with cytosines to amplify methylated DNA, and one set recognizes DNA with uracil or thymine to amplify unmethylated DNA. MSP using qPCR can also be performed to obtain quantitative rather than qualitative information about methylation.
- Miniprimer PCR: uses a thermostable polymerase (S-Tbr) that can extend from short primers ("smalligos") as short as 9 or 10 nucleotides. This method permits PCR targeting to smaller primer binding regions, and is used to amplify conserved DNA sequences, such as the 16S (or eukaryotic 18S) rRNA gene.[45]
- Multiplex ligation-dependent probe amplification (MLPA): permits amplifying multiple targets with a single primer pair, thus avoiding the resolution limitations of multiplex PCR (see below).
- Multiplex-PCR: consists of multiple primer sets within a single PCR mixture to produce amplicons of varying sizes that are specific to different DNA sequences. By targeting multiple genes at once, additional information may be gained from a single test-run that otherwise would require several times the reagents and more time to perform. Annealing temperatures for each of the primer sets must be optimized to work correctly within a single reaction, and amplicon sizes. That is, their base pair length should be different enough to form distinct bands when visualized by gel electrophoresis.
- Nanoparticle-Assisted PCR (nanoPCR): In recent years, it has been reported that some nanoparticles (NPs) can enhance the efficiency of PCR (thus being called nanoPCR), and some even perform better than the original PCR enhancers. It was also found that quantum dots (QDs) can improve PCR specificity and efficiency. Single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs) are efficient in enhancing the amplification of long PCR. Carbon nanopowder (CNP) was reported be able to improve the efficiency of repeated PCR and long PCR. ZnO, TiO2, and Ag NPs were also found to increase PCR yield. Importantly, already known data has indicated that non-metallic NPs retained acceptable amplification fidelity. Given that many NPs are capable of enhancing PCR efficiency, it is clear that there is likely to be great potential for nanoPCR technology improvements and product development.[46][47]
- Nested PCR: increases the specificity of DNA amplification, by reducing background due to non-specific amplification of DNA. Two sets of primers are used in two successive PCRs. In the first reaction, one pair of primers is used to generate DNA products, which besides the intended target, may still consist of non-specifically amplified DNA fragments. The product(s) are then used in a second PCR with a set of primers whose binding sites are completely or partially different from and located 3' of each of the primers used in the first reaction. Nested PCR is often more successful in specifically amplifying long DNA fragments than conventional PCR, but it requires more detailed knowledge of the target sequences.
- Overlap-extension PCR or Splicing by overlap extension (SOEing) : a genetic engineering technique that is used to splice together two or more DNA fragments that contain complementary sequences. It is used to join DNA pieces containing genes, regulatory sequences, or mutations; the technique enables creation of specific and long DNA constructs. It can also introduce deletions, insertions or point mutations into a DNA sequence.[48][49]
- PAN-AC: uses isothermal conditions for amplification, and may be used in living cells.[50][51]
- quantitative PCR (qPCR): used to measure the quantity of a target sequence (commonly in real-time). It quantitatively measures starting amounts of DNA, cDNA, or RNA. quantitative PCR is commonly used to determine whether a DNA sequence is present in a sample and the number of its copies in the sample. Quantitative PCR has a very high degree of precision. Quantitative PCR methods use fluorescent dyes, such as Sybr Green, EvaGreen or fluorophore-containing DNA probes, such as TaqMan, to measure the amount of amplified product in real time. It is also sometimes abbreviated to RT-PCR (real-time PCR) but this abbreviation should be used only for reverse transcription PCR. qPCR is the appropriate contractions for quantitative PCR (real-time PCR).
- Reverse Transcription PCR (RT-PCR): for amplifying DNA from RNA. Reverse transcriptase reverse transcribes RNA into cDNA, which is then amplified by PCR. RT-PCR is widely used in expression profiling, to determine the expression of a gene or to identify the sequence of an RNA transcript, including transcription start and termination sites. If the genomic DNA sequence of a gene is known, RT-PCR can be used to map the location of exons and introns in the gene. The 5' end of a gene (corresponding to the transcription start site) is typically identified by RACE-PCR (Rapid Amplification of cDNA Ends).
- Solid Phase PCR: encompasses multiple meanings, including Polony Amplification (where PCR colonies are derived in a gel matrix, for example), Bridge PCR[52] (primers are covalently linked to a solid-support surface), conventional Solid Phase PCR (where Asymmetric PCR is applied in the presence of solid support bearing primer with sequence matching one of the aqueous primers) and Enhanced Solid Phase PCR[53] (where conventional Solid Phase PCR can be improved by employing high Tm and nested solid support primer with optional application of a thermal 'step' to favour solid support priming).
- Suicide PCR: typically used in paleogenetics or other studies where avoiding false positives and ensuring the specificity of the amplified fragment is the highest priority. It was originally described in a study to verify the presence of the microbe Yersinia pestis in dental samples obtained from 14th Century graves of people supposedly killed by plague during the medieval Black Death epidemic.[54] The method prescribes the use of any primer combination only once in a PCR (hence the term "suicide"), which should never have been used in any positive control PCR reaction, and the primers should always target a genomic region never amplified before in the lab using this or any other set of primers. This ensures that no contaminating DNA from previous PCR reactions is present in the lab, which could otherwise generate false positives.
- Thermal asymmetric interlaced PCR (TAIL-PCR): for isolation of an unknown sequence flanking a known sequence. Within the known sequence, TAIL-PCR uses a nested pair of primers with differing annealing temperatures; a degenerate primer is used to amplify in the other direction from the unknown sequence.[55]
- Touchdown PCR (Step-down PCR): a variant of PCR that aims to reduce nonspecific background by gradually lowering the annealing temperature as PCR cycling progresses. The annealing temperature at the initial cycles is usually a few degrees (3–5 °C) above the Tm of the primers used, while at the later cycles, it is a few degrees (3–5 °C) below the primer Tm. The higher temperatures give greater specificity for primer binding, and the lower temperatures permit more efficient amplification from the specific products formed during the initial cycles.[56]
- Universal Fast Walking: for genome walking and genetic fingerprinting using a more specific 'two-sided' PCR than conventional 'one-sided' approaches (using only one gene-specific primer and one general primer—which can lead to artefactual 'noise')[57] by virtue of a mechanism involving lariat structure formation. Streamlined derivatives of UFW are LaNe RAGE (lariat-dependent nested PCR for rapid amplification of genomic DNA ends),[58] 5'RACE LaNe[59] and 3'RACE LaNe.[60]
History
A 1971 paper in the Journal of Molecular Biology by Kjell Kleppe and co-workers in the laboratory of H. Gobind Khorana first described a method using an enzymatic assay to replicate a short DNA template with primers in vitro.[61] However, this early manifestation of the basic PCR principle did not receive much attention at the time, and the invention of the polymerase chain reaction in 1983 is generally credited to Kary Mullis.[62]
%2C_c_1986._(9663810586).jpg)
When Mullis developed the PCR in 1983, he was working in Emeryville, California for Cetus Corporation, one of the first biotechnology companies. There, he was responsible for synthesizing short chains of DNA. Mullis has written that he conceived of PCR while cruising along the Pacific Coast Highway one night in his car.[63] He was playing in his mind with a new way of analyzing changes (mutations) in DNA when he realized that he had instead invented a method of amplifying any DNA region through repeated cycles of duplication driven by DNA polymerase. In Scientific American, Mullis summarized the procedure: "Beginning with a single molecule of the genetic material DNA, the PCR can generate 100 billion similar molecules in an afternoon. The reaction is easy to execute. It requires no more than a test tube, a few simple reagents, and a source of heat."[64] He was awarded the Nobel Prize in Chemistry in 1993 for his invention,[6] seven years after he and his colleagues at Cetus first put his proposal to practice. However, some controversies have remained about the intellectual and practical contributions of other scientists to Mullis' work, and whether he had been the sole inventor of the PCR principle (see below).
At the core of the PCR method is the use of a suitable DNA polymerase able to withstand the high temperatures of >90 °C (194 °F) required for separation of the two DNA strands in the DNA double helix after each replication cycle. The DNA polymerases initially employed for in vitro experiments presaging PCR were unable to withstand these high temperatures.[4] So the early procedures for DNA replication were very inefficient and time-consuming, and required large amounts of DNA polymerase and continuous handling throughout the process.
The discovery in 1976 of Taq polymerase — a DNA polymerase purified from the thermophilic bacterium, Thermus aquaticus, which naturally lives in hot (50 to 80 °C (122 to 176 °F)) environments[14] such as hot springs — paved the way for dramatic improvements of the PCR method. The DNA polymerase isolated from T. aquaticus is stable at high temperatures remaining active even after DNA denaturation,[15] thus obviating the need to add new DNA polymerase after each cycle.[5] This allowed an automated thermocycler-based process for DNA amplification.
Patent disputes
The PCR technique was patented by Kary Mullis and assigned to Cetus Corporation, where Mullis worked when he invented the technique in 1983. The Taq polymerase enzyme was also covered by patents. There have been several high-profile lawsuits related to the technique, including an unsuccessful lawsuit brought by DuPont. The pharmaceutical company Hoffmann-La Roche purchased the rights to the patents in 1992 and currently holds those that are still protected.
A related patent battle over the Taq polymerase enzyme is still ongoing in several jurisdictions around the world between Roche and Promega. The legal arguments have extended beyond the lives of the original PCR and Taq polymerase patents, which expired on March 28, 2005.[65]
References
- 1 2 "PCR".
- ↑ Bartlett, J. M. S.; Stirling, D. (2003). "A Short History of the Polymerase Chain Reaction". PCR Protocols. Methods in Molecular Biology. 226 (2nd ed.). pp. 3–6. doi:10.1385/1-59259-384-4:3. ISBN 1-59259-384-4.
- ↑ "Process for amplifying, detecting, and/or-cloning nucleic acid sequences".
- 1 2 3 Saiki, R.; Scharf, S.; Faloona, F.; Mullis, K.; Horn, G.; Erlich, H.; Arnheim, N. (1985). "Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia". Science. 230 (4732): 1350–1354. doi:10.1126/science.2999980. PMID 2999980.
- 1 2 Saiki, R.; Gelfand, D.; Stoffel, S.; Scharf, S.; Higuchi, R.; Horn, G.; Mullis, K.; Erlich, H. (1988). "Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase". Science. 239 (4839): 487–491. doi:10.1126/science.2448875. PMID 2448875.
- 1 2 "Kary B. Mullis - Nobel Lecture: The Polymerase Chain Reaction".
- ↑ Cheng, S.; Fockler, C.; Barnes, W. M.; Higuchi, R. (1994). "Effective Amplification of Long Targets from Cloned Inserts and Human Genomic DNA". Proceedings of the National Academy of Sciences. 91 (12): 5695–5699. doi:10.1073/pnas.91.12.5695. PMC 44063
 . PMID 8202550.
. PMID 8202550. - ↑ Carr AC, Moore SD (2012). Lucia, Alejandro, ed. "Robust quantification of polymerase chain reactions using global fitting". PLOS ONE. 7 (5): e37640. doi:10.1371/journal.pone.0037640. PMC 3365123. PMID 22701526.
- 1 2 Joseph Sambrook & David W. Russel (2001). Molecular Cloning: A Laboratory Manual (3rd ed.). Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. ISBN 0-879-69576-5. Chapter 8: In vitro Amplification of DNA by the Polymerase Chain Reaction
- ↑ "Polymerase Chain Reaction (PCR)".
- ↑ Pavlov, A. R.; Pavlova, N. V.; Kozyavkin, S. A.; Slesarev, A. I. (2004). "Recent developments in the optimization of thermostable DNA polymerases for efficient applications☆". Trends in Biotechnology. 22 (5): 253–260. doi:10.1016/j.tibtech.2004.02.011. PMID 15109812.
- ↑ Rychlik W, Spencer WJ, Rhoads RE (1990). "Optimization of the annealing temperature for DNA amplification in vitro". Nucl Acids Res. 18 (21): 6409–6412. doi:10.1093/nar/18.21.6409. PMC 332522. PMID 2243783.
- 1 2 Sharkey, D. J.; Scalice, E. R.; Christy, K. G.; Atwood, S. M.; Daiss, J. L. (1994). "Antibodies as Thermolabile Switches: High Temperature Triggering for the Polymerase Chain Reaction". Bio/Technology. 12 (5): 506–509. doi:10.1038/nbt0594-506.
- 1 2 Chien A, Edgar DB, Trela JM (1976). "Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus". J. Bacteriol. 127 (3): 1550–1557. PMC 232952. PMID 8432.
- 1 2 Lawyer, F.; Stoffel, S.; Saiki, R.; Chang, S.; Landre, P.; Abramson, R.; Gelfand, D. (1993). "High-level expression, purification, and enzymatic characterization of full-length Thermus aquaticus DNA polymerase and a truncated form deficient in 5' to 3' exonuclease activity". PCR methods and applications. 2 (4): 275–287. doi:10.1101/gr.2.4.275. PMID 8324500.
- ↑ https://bbhosted.cuny.edu/bbcswebdav/pid-23905396-dt-content-rid-116035749_1/courses/HTR01_CHEM_37800_01_1162_1/Recitation%209%20PCR.pdf
- ↑ Campbell Biology,7th edition
- ↑ PCR from problematic templates. Focus 22:1 p.10 (2000).
- ↑ Helpful tips for PCR. Focus 22:1 p.12 (2000).
- ↑ Sarkar, G.; Kapelner, S.; Sommer, S. (1990). "Formamide can dramatically improve the specificity of PCR". Nucleic Acids Research. 18 (24): 7465. doi:10.1093/nar/18.24.7465. PMC 332902. PMID 2259646.
- ↑ "Electronic PCR". NCBI – National Center for Biotechnology Information. Retrieved 13 March 2012.
- ↑ Pavlov AR, Pavlova NV, Kozyavkin SA, Slesarev AI (2006). "Thermostable DNA Polymerases for a Wide Spectrum of Applications: Comparison of a Robust Hybrid TopoTaq to other enzymes". In Kieleczawa J. DNA Sequencing II: Optimizing Preparation and Cleanup. Jones and Bartlett. pp. 241–257. ISBN 0-7637-3383-0.
- ↑ Pombert JF, Sistek V, Boissinot M, Frenette M (2009). "Evolutionary relationships among salivarius streptococci as inferred from multilocus phylogenies based on 16S rRNA-encoding, recA, secA, and secY gene sequences". BMC Microbiol. 9: 232. doi:10.1186/1471-2180-9-232. PMC 2777182. PMID 19878555.
- ↑ "Chemical Synthesis, Sequencing, and Amplification of DNA (class notes on MBB/BIO 343)". Arizona State University. Retrieved 2007-10-29.
- ↑ Quill E "Blood-Matching Goes Genetic" Science Magazine (14 March 2008) pp. 1478-1479.
- ↑ Tomar, Rukam (2010). Molecular Markers and Plant Biotechnology. Pitman Pura, New Delhi: New India Publishing Agency. p. 188. ISBN 978-93-80235-25-7.
- 1 2 3 Cai, H; Caswell JL; Prescott JF (March 2014). "Nonculture Molecular Techniques for Diagnosis of Bacterial Disease in Animals: A Diagnostic Laboratory Perspective". Veterinary Pathology. 51 (2): 341–350. doi:10.1177/0300985813511132. PMID 24569613.
- ↑ Salis AD (2009). "Applications in Clinical Microbiology". Real-Time PCR: Current Technology and Applications. Caister Academic Press. ISBN 978-1-904455-39-4.
- ↑ Kwok S et al. "Identification of HIV sequences by using in vitro enzymatic amplification and oligomer cleavage detection." J. Virol. vol. 61(5) pp. 1690-4 (1987).
- ↑ Boehnke M et al. "Fine-structure genetic mapping of human chromosomes using the polymerase chain reaction on single sperm." Am J Hum Genet vol. 45(1) pp. 21-32 (1989).
- ↑ Garibyan L, Avashia N (2013). "Polymerase Chain Reaction". Journal of Investigative Dermatology. 133: 1–4. doi:10.1038/jid.2013.1.
- ↑ Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC, Markham AF (1989). "Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS)". Nucleic Acids Research. 17 (7): 2503–2516. doi:10.1093/nar/17.7.2503. PMC 317639. PMID 2785681.
- ↑ Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL (1995). "Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides". Gene. 164 (1): 49–53. doi:10.1016/0378-1119(95)00511-4. PMID 7590320.
- ↑ Innis MA, Myambo KB, Gelfand DH, Brow MA (1988). "DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA". Proc Natl Acad Sci USA. 85 (24): 9436–4940. doi:10.1073/pnas.85.24.9436. PMC 282767. PMID 3200828.
- ↑ Pierce KE & Wangh LJ (2007). "Linear-after-the-exponential polymerase chain reaction and allied technologies Real-time detection strategies for rapid, reliable diagnosis from single cells". Methods Mol Med. Methods in Molecular Medicine™. 132: 65–85. doi:10.1007/978-1-59745-298-4_7. ISBN 978-1-58829-578-1. PMID 17876077.
- ↑ Schwartz JJ, Lee C, Shendure J (2012). "Accurate gene synthesis with tag-directed retrieval of sequence-verified DNA molecules". Nature Methods. 9 (9): 913–915. doi:10.1038/nmeth.2137. PMC 3433648. PMID 22886093.
- ↑ Vincent M, Xu Y, Kong H (2004). "Helicase-dependent isothermal DNA amplification". EMBO Reports. 5 (8): 795–800. doi:10.1038/sj.embor.7400200. PMC 1249482. PMID 15247927.
- ↑ Chou Q, Russell M, Birch DE, Raymond J, Bloch W (1992). "Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications". Nucleic Acids Research. 20 (7): 1717–1723. doi:10.1093/nar/20.7.1717. PMC 312262. PMID 1579465.
- ↑ Kellogg, DE; Rybalkin, I; Chen, S; Mukhamedova, N; Vlasik, T; Siebert, PD; Chenchik, A (1994). "TaqStart Antibody: "hot start" PCR facilitated by a neutralizing monoclonal antibody directed against Taq DNA polymerase". BioTechniques. 16 (6): 1134–7. PMID 8074881.
- ↑ San Millan RM, Martinez-Ballesteros I, Rementeria A, Garaizar J, Bikandi J (2013). "Online exercise for the design and simulation of PCR and PCR-RFLP experiments". BMC Research Notes. 6: 513. doi:10.1186/1756-0500-6-513. PMID 24314313.
- ↑ E. Zietkiewicz; A. Rafalski & D. Labuda (1994). "Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification". Genomics. 20 (2): 176–83. doi:10.1006/geno.1994.1151. PMID 8020964.
- ↑ Ochman H, Gerber AS, Hartl DL (1988). "Genetic Applications of an Inverse Polymerase Chain Reaction". Genetics. 120 (3): 621–623. PMC 1203539. PMID 2852134.
- ↑ Mueller PR, Wold B (1988). "In vivo footprinting of a muscle specific enhancer by ligation mediated PCR". Science. 246 (4931): 780–786. doi:10.1126/science.2814500. PMID 2814500.
- ↑ Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (1996). "Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands". Proc Natl Acad Sci USA. 93 (13): 9821–9826. doi:10.1073/pnas.93.18.9821. PMC 38513. PMID 8790415.
- ↑ Isenbarger TA, Finney M, Ríos-Velázquez C, Handelsman J, Ruvkun G (2008). "Miniprimer PCR, a New Lens for Viewing the Microbial World". Applied and Environmental Microbiology. 74 (3): 840–9. doi:10.1128/AEM.01933-07. PMC 2227730. PMID 18083877.
- ↑ Cenchao Shen; Wenjuan Yang; Qiaoli Ji; Hisaji Maki; Anjie Dong; Zhizhou Zhang (2009). "NanoPCR observation: different levels of DNA replication fidelity in nanoparticle-enhanced polymerase chain reactions". Nanotechnology. 20: 455103. doi:10.1088/0957-4484/20/45/455103.
- ↑ Shen, Cenchao (2013). An Overview of Nanoparticle‐Assisted Polymerase Chain Reaction Technology. US: Wiley-Blackwell Publishing Ltd. pp. 97–106.
- ↑ Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR (1989). "Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap exten-sion". Gene. 77 (1): 61–68. doi:10.1016/0378-1119(89)90359-4. PMID 2744488.
- ↑ Moller, Simon (2006). PCR (THE BASICS). US: Taylor & Francis Group. p. 144.
- ↑ David F, Turlotte E (1998). "Une méthode d'amplification génique isotherme" [An Isothermal Amplification Method]. Comptes Rendus de l'Académie des Sciences - Series III - Sciences de la Vie. 321 (11): 909–914. doi:10.1016/S0764-4469(99)80005-5. ISSN 0764-4469.
- ↑ Fabrice David (September–October 2002). "Utiliser les propriétés topologiques de l'ADN: une nouvelle arme contre les agents pathogènes" (PDF). Fusion.(in French)
- ↑ Bing DH, Boles C, Rehman FN, Audeh M, Belmarsh M, Kelley B, Adams CP (1996). "Bridge amplification: a solid phase PCR system for the amplification and detection of allelic differences in single copy genes". Genetic Identity Conference Proceedings, Seventh International Symposium on Human Identification.
- ↑ Khan Z, Poetter K, Park DJ (2008). "Enhanced solid phase PCR: mechanisms to increase priming by solid support primers". Analytical Biochemistry. 375 (2): 391–393. doi:10.1016/j.ab.2008.01.021. PMID 18267099.
- ↑ Raoult, D; G Aboudharam; E Crubezy; G Larrouy; B Ludes; M Drancourt (2000-11-07). "Molecular identification by "suicide PCR" of Yersinia pestis as the agent of medieval black death". Proc. Natl. Acad. Sci. U.S.A. 97 (23): 12800–12803. doi:10.1073/pnas.220225197. ISSN 0027-8424. PMC 18844. PMID 11058154.
- ↑ Y.G. Liu & R. F. Whittier (1995). "Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking". Genomics. 25 (3): 674–81. doi:10.1016/0888-7543(95)80010-J. PMID 7759102.
- ↑ Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (1991). "'Touchdown' PCR to circumvent spurious priming during gene amplification". Nucl Acids Res. 19 (14): 4008. doi:10.1093/nar/19.14.4008. PMC 328507. PMID 1861999.
- ↑ Myrick KV, Gelbart WM (2002). "Universal Fast Walking for direct and versatile determination of flanking sequence". Gene. 284 (1–2): 125–131. doi:10.1016/S0378-1119(02)00384-0. PMID 11891053.
- ↑ "Full Text - LaNe RAGE: a new tool for genomic DNA flanking sequence determination".
- ↑ Park DJ (2005). "A new 5' terminal murine GAPDH exon identified using 5'RACE LaNe". Molecular Biotechnology. 29 (1): 39–46. doi:10.1385/MB:29:1:39. PMID 15668518.
- ↑ Park DJ (2004). "3'RACE LaNe: a simple and rapid fully nested PCR method to determine 3'-terminal cDNA sequence". Biotechniques. 36 (4): 586–588, 590. PMID 15088375.
- ↑ Kleppe K, Ohtsuka E, Kleppe R, Molineux I, Khorana HG (1971). "Studies on polynucleotides. XCVI. Repair replications of short synthetic DNA's as catalyzed by DNA polymerases". J. Mol. Biol. 56 (2): 341–361. doi:10.1016/0022-2836(71)90469-4. PMID 4927950.
- ↑ Rabinow, Paul (1996). Making PCR: A Story of Biotechnology. Chicago: University of Chicago Press. ISBN 0-226-70146-8.
- ↑ Mullis, Kary (1998). Dancing Naked in the Mind Field. New York: Pantheon Books. ISBN 0-679-44255-3.
- ↑ Mullis, Kary (1990). "The unusual origin of the polymerase chain reaction". Scientific American. 262 (4): 56–61, 64–5. doi:10.1038/scientificamerican0490-56. PMID 2315679.
- ↑ Advice on How to Survive the Taq Wars ¶2: GEN Genetic Engineering News - Biobusiness Channel: Article. May 1, 2006 (Vol. 26, No. 9).
External links
| Library resources about Polymerase chain reaction |
| Wikimedia Commons has media related to Polymerase chain reaction. |
- A Guide to PCR Technologies SelectScience
- OpenPCR Open-source PCR thermalcycler project
- US Patent for PCR
- Step-through animation of PCR – Cold Spring Harbor Laboratory
- OpenWetWare
- What is PCR plateau effect? YouTube tutorial video
- GeneWarrior Online PCR Primer design tool
- History of the Polymerase Chain Reaction from the Smithsonian Institution Archives
- 3d models of PCR equipment for 3D printing on thingiverse.com
- Computer exercise. Design of PCR and PCR-RFLP experiments