Thermal shift assay
A thermal shift assay quantifies the change in thermal denaturation temperature of a protein under varying conditions. The differing conditions that can be examined are very diverse, e.g. pH, salts, additives, drugs, drug leads, oxidation/reduction, or mutations. The binding of low molecular weight ligands can increase the thermal stability of a protein, as described by Koshland (1958)[1] and Linderstrom-Lang and Schellman (1959).[2] Almost half of enzymes require a metal ion co-factor.[3] Thermostable proteins are often more useful than their non-thermostable counterparts, e.g. DNA polymerase in the polymerase chain reaction,[4] so protein engineering often includes adding mutations to increase thermal stability. Protein crystallisation is more successful for proteins with a higher melting point[5] and adding buffer components that stabilise proteins improve the likelihood of protein crystals forming.[6] If examining pH then the possible effects of the buffer molecule on thermal stability should be taken into account along with the fact that pKa of each buffer molecule changes uniquely with temperature.[7] Additionally, any time a charged species is examined the effects of the counterion should be accounted for.
Thermal stability of proteins has traditionally been investigated using biochemical assays, circular dichroism, or differential scanning calorimetry. Biochemical assays require a catalytic activity of the protein in question as well as a specific assay. Circular dichroism and differential scanning calorimetry both consume large amounts of protein and are low-throughput methods. The thermofluor assay was the first high-throughput thermal shift assay and its utility and limitations has spurred the invention of a plethora of alternate methods. Each method has its strengths and weaknesses but they all struggle with intrinsically disordered proteins without any clearly defined tertiary structure as the essence of a thermal shift assay is measuring the temperature at which a protein goes from well-defined structure to disorder.
Methods
Thermofluor
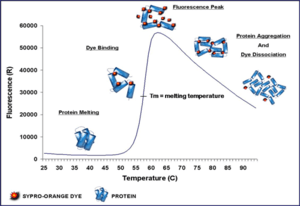
The technique was first described by Semisotnov et al. (1991)[8] using 1,8-ANS and quartz cuvettes. 3 Dimensional Pharmaceuticals were the first to describe a high-throughput version using a plate reader[9] and Wyeth Research published a variation of the method with SYPRO Orange instead of 1,8-ANS.[10] SYPRO Orange has an excitation/emission wavelength profile compatible with qPCR machines which are almost ubiquitous in institutions that perform molecular biology research. The name Differential Scanning Fluorimetry (DSF) was introduced later[11] but thermofluor is preferable as thermofluor is no longer trademarked and differential scanning fluorimetry is easily confused with differential scanning calorimetry.
SYPRO Orange binds nonspecifically to hydrophobic surfaces, and water strongly quenches its fluorescence. When the protein unfolds, the exposed hydrophobic surfaces bind the dye, resulting in an increase in fluorescence by excluding water. Detergent micelles will also bind the dye and increase background noise dramatically - this effect is lessened by switching to the dye ANS[12] but that requires UV excitation. The stability curve and its midpoint value (melting temperature, Tm also known as the temperature of hydrophobic exposure, Th) are obtained by gradually increasing the temperature to unfold the protein and measuring the fluorescence at each point. Curves are measured for protein only and protein + ligand, and ΔTm is calculated. The method might not work very well for protein-protein interactions if one of the interaction partners contains large hydrophobic patches as it is highly non-trivial to dissect prevention of aggregation, stabilisation of a native folds and steric hindrance of dye access to hydrophobic sites. In addition, partly aggregated protein can also limit the relative fluorescence increase upon heating; in extreme cases there will be no fluorescence increase at all because all protein is already in aggregates before heating. Knowing this effect can be very useful as a high relative fluorescence increase suggests a significant fraction of folded protein in the starting material.
This assay allows high-throughput screening of ligands to the target protein and it is widely used in the early stages of drug discovery in the pharmaceutical industry, structural genomics efforts, and high-throughput protein engineering.[13]
A typical assay
- Materials: A fluorometer equipped with temperature control or similar instrumentation (RT-PCR machines); suitable fluorescent dye; a suitable assay plate, such as 96 well RT-PCR plate.
- Compound solutions: Test ligands are prepared at a 50- to 100-fold concentrated solution, generally in the 10-100 mM range. For titration, a typical experimental protocol employs a set of 12 well, comprising 11 different concentrations of a test compound with a single negative control well.
- Protein solution: Typically, target protein is diluted from a concentrated stock to a working concentration of ~0.5-5 μM protein with dye into a suitable assay buffer. The exact concentrations of protein and dye are defined by experimental assay development studies.
- Centrifugation and oil dispense: A brief centrifugation (~1000 × g-force, 1 min) of the assay plate to mix compounds into the protein solution, 1-2 μl silicone oil to prevent the evaporation during heating is overlaid onto the solution (some systems use plastic seals instead), followed by an additional centrifugation step (~1000 × g-force, 1 min).
- Instrumental set up: A typical temperature ramp rates range from 0.1-10 °C/min but generally in the range of 1 °C/min. The fluorescence in each well is measured at regular intervals, 0.2-1 °C/image, over a temperature range spanning the typical protein unfolding temperatures of 25-95 °C.[14]
CPM, thiol-specific dyes
Alexandrov et al. (2008)[15] published a variation on the thermofluor assay where SYPRO Orange was replaced by N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM), a compound that only fluoresces after reacting with a nucleophile. CPM has a high preference for thiols over other typical biological nucleophiles and therefore will react with cysteine side chains before others. Cysteines are typically buried in the interior of a folded protein as they are hydrophobic. When a protein denatures cysteine thiols become available and a fluorescent signal can be read from reacted CPM. The exitation and emission wavelengths for reacted CPM are 387 nm/ 463 nm so a fluorescence plate reader or a qPCR machine with specialised filters is required. Alexandrov et al. used the technique successfully on the membrane proteins Apelin GPCR and FAAH as well as β-lactoglobin which fibrillates on heating rather than going to a molten globule.
DCVJ, rigidity sensitive dyes
4-(dicyanovinyl)julolidine (DCVJ) is a molecular rotor probe with fluorescence that is strongly dependent on the rigidity of its environment. When protein denatures DCVJ increases in fluorescence. It has been reported to work with 40 mg/ml of antibody.[16]
Intrinsic tryptophan fluorescence lifetime
The lifetime of tryptophan fluorescence differs between folded and unfolded protein and by measuring the lifetime of UV-excited fluorescence at temperature intervals you can get a measurement of the melting temperature of your protein.
A prominent advantage is that no reporter dyes are required to be added as tryptophan is an intrinsic part of the protein. This can also be a disadvantage as not all proteins contain tryptophan. Intrinsic fluorescence lifetime works with membrane proteins and detergent micelles but a powerful UV fluorescer in the buffer could drown out the signal and few articles are published using the technique for thermal shift assays.
Intrinsic tryptophan fluorescence wavelength
The excitation and emission wavelengths of tryptophan are dependent on the immediate environment and therefore differs between folded and unfolded protein, just as the fluorescence lifetime. Currently there are at least two machines on the market that can read this shift in wavelength in a high-throughput manner while heating the samples.
The advantages and disadvantages are the same as for fluorescence lifetime except that there are more examples in the scientific literature of use.
Static light scattering
Static light scattering allows monitoring of the sizes of the species in solution. Since proteins typically aggregate upon denaturation (or form fibrils) the detected species size will go up.
This is label-free and independent of specific residues in the protein or buffer composition. The only requirement is that the protein actually aggregates/fibrillates after denaturation and that the protein of interest has been purified.
FastPP
In fast parallel proteolysis the researcher adds a thermostable protease (thermolysin) and takes out samples in parallel upon heating in a thermal gradient cycler.[17] Optionally, for instance for lowly expressed proteins, a Western blot is then run to determine at what temperature a protein becomes degraded. For pure or highly enriched proteins, direct SDS-PAGE detection is possible facilitating Commassie-fluorescence based direct quantification. FastPP exploits that proteins become increasingly susceptible to proteolysis when unfolded and that thermolysin cleaves at hydrophobic residues which are typically found in the core of proteins.
To reduce the workload, Western Blots could be replaced by SDS-PAGE gel polyhistidine-tag staining, provided that the protein has such a tag and is expressed in adequate amounts.
FastPP can be used on unpurified, complex mixtures of proteins and proteins fused with other proteins, such as GST or GFP, as long as the sequence that is the target of the Western blot, e.g. His-tag, is directly linked to the protein of interest. However, commercially available thermolysin is dependent on calcium ions for activity and denatures itself just above 85 degrees Celsius. So calcium must be present and calcium chelators absent in the buffer - other compounds that interfere with the function (such as high concentrations of detegents) of the protease could also be problematic.
FASTpp has also been used to monitor binding-coupled folding of intrinsically disordered proteins (IDPs).
CETSA
CETSA stands for cellular thermal shift assay.[18] Very similar to fastPP but instead of adding a protease samples are centrifuged after heating and the supernatant is run on Western blots or two target-directed antibodies are added and their relative proximity is detected through FRET. When a protein denatures it aggregates and becomes part of the pellet and can therefore not be found in the supernatant. An intrinsic limitation of this assay is that not all proteins aggregate upon unfolding but might instead populate highly soluble, relatively compact molten-globule (like) conformations. In addition, it is possible that proteins will co-precipitate with their less stable protein interaction partners and therefore show lower apparent stability than their actual thermodynamic stability.
Just as for FastPP the Western blot could be replaced by staining for a polyhistidine-tag in the SDS-PAGE gel. Additionally, instead of using two antibodies the FRET method could be performed with a single fluorescent antibody and a fluorescently labelled peptide recognised by the antibody.[19]
A technique that has been shown to work on both complex mixtures and whole cells but unfortunately requires antibodies. The protein of interest should not be fused to other proteins such as MBP or GFP as they could keep a denatured protein in solution.
A CETSA assay for membrane proteins was described recently. This method not only allows to screen ligands but also thermostabilizing conditions.[20]
ThermoFAD
Thermofluor variant specific for flavin-binding proteins. Analogous to thermofluor binding assays, a small volume of protein solution is heated up and the fluorescence increase is followed as function of temperature. In contrast to Thermofluor, no external fluorescent dye is needed because the flavin cofactor is already present in the flavin-binding protein and its fluorescence properties change upon unfolding. [21]
SEC-TS
Size exclusion chromatography can be used directly to access protein stability in the presence or absence of ligands.[22] Samples of purified protein are heated in a water bath or thermocycler, cooled, centrifuged to remove aggregated proteins, and run on an analytical HPLC. As the melting temperature is reached and protein precipitates or aggregates, peak height decreases and void peak height increases. This can be used to identify ligands and inhibitors, and optimize purification conditions.[23][24]
While of lower through-put than FSEC-TS, requiring large amounts of purified protein, SEC-TS avoids any influence of the fluorescent tag on apparent protein stability.
FSEC-TS
In fluorescence-detection size exclusion chromatography the protein of interest is fluorescently tagged, e.g. with GFP, and run through a gel filtration column on an FPLC system equipped with a fluorescence detector. The resulting chromatogram allows allows the researcher to estimate the monodispersity and expression level of the tagged protein in the current buffer.[25] Since only fluorescence is measured, only the tagged protein is seen in the chromatogram. FSEC is typically used to compare membrane protein orthologs or screen detergents to solubilise specific membrane proteins in.
For fluorescence-detection size-exclusion chromatography-based thermostability assay (FSEC-TS) the samples are heated in the same manner as in FastPP and CETSA and after centrifugation to clear away precipitate the supernatant is treated in the same manner as FSEC.[26] Larger aggregates are seen in the void volume while the peak height for the protein of interest decreases when the unfolding temperature is reached.
GFP has a Tm of ~76 C so the technique is limited to temperature below ~70 C. Of all the techniques mentioned it may have the lowest throughput but it is free of antibody requirements and works on complex mixtures of proteins.
Radioligand binding thermostability assay
GPCRs are pharmacologically important transmembrane proteins. Their x-ray crystal structures were revealed long after other transmembrane proteins of lesser interest. The difficulty in obtaining protein crystals of GPCRs was likely due to their high flexibility. Less flexible versions were obtained by truncating, mutating, and inserting T4 lysozyme in the recombinant sequence. One of the methods researchers used to guide these alterations was radioligand binding thermostability assay.[27]
The assay is performed by incubating the protein with a radiolabelled ligand of the protein for 30 minutes at a given temperature, then quench on ice, run through a gel filtration mini column, and quantify the radiation levels of the protein that comes off the column. The radioligand concentration is high enough to saturate the protein. Denatured protein is unable to bind the radioligand and the protein and radioligand will be separated in the gel filtration mini column. When screening mutants selection will be for thermal stability in the specific conformation, i.e. if the radioligand is an agonist selection will be for the agonist binding conformation and if it is an antagonist then the screening is for stability in the antagonist binding conformation.
Radioassays have the advantage of working with minute amounts of protein. But it is work with radioactive substances and large amount of manual labour is involved. A high-affinity ligand has to be known for the protein of interest and the buffer must not interfere with the binding of the radioligand. Other thermal shift assays can also select for specific conformations if a ligand of the appropriate type is added to the experiment.
Comparisons of the various approaches
| Thermofluor | CPM | DCVJ | Tryptophan fluorescence lifetime | Tryptophan fluorescence wavelength | Static light scattering | FastPP | CETSA | SEC-TS | FSEC-TS | Radioligand binding thermostability assay | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Purification | Pure protein | Pure protein | Pure protein at very high concentration | Pure protein | Pure protein | Pure protein | Complex mixture | Complex mixture | Pure protein | Complex mixture | Complex mixture |
| Detergents | Below cmc | Yes | Yes | Yes | Yes | Yes | Yes (low conc.) | Yes | Yes | Yes | Yes |
| Buffer | Avoid very high conc. organic solvents | Avoid thiols, possibly other nucleophiles | - | - | - | - | Thermolysin requires calcium ions | - | - | - | Should maximize radioligand binding affinity |
| Throughput | Highest | Highest | Highest | Intermediate | Intermediate | Intermediate | Intermediate-low (if western required) | Lowest (intermediate with FRET) | Lowest | Lowest | Lowest |
| Sequence requirements | No | Cysteines, not on surface and not in disulphide bonds | No | Tryptophan | Tryptophan | No | Sequence must contain hydrophobic residues (required for cleavage) | For antibody | No | For fluorescent tag | No |
| Equipment requirements | qPCR machine | qPCR machine with UV excitation or fluorescence plate reader | qPCR machine | High-throughput differential intrinsic fluorescence lifetime reader | High-throughput differential intrinsic fluorescence wavelength reader | High-throughput differential static light scattering reader | Thermal cycler (or water bath) and Western blot | Thermal cycler (or water bath) and Western blot (or FRET reader) | Thermal cycler (or water bath) and HPLC | Thermal cycler (or water bath) and FPLC with fluorescence reader | Thermal cycler (or water bath) and scintillation counter |
| Labels | Yes + DMSO | Yes + DMSO | Yes + DMSO | Label-free | Label-free | Label-free | Label-free | Label-free | Label-free | Yes | Radioligand |
| Possible interference from fluorophores in buffer | Yes | Yes | Yes | Yes | Yes | No | No | No | No | Yes | No |
| Fusion proteins | No | Possibly | No | Possibly | Possibly | No | Optional | No | No | Yes | Yes |
| Works with proteins that fibrillate when heated | No | Yes, at least with beta-lactoglobulin | Yes | Yes, probably | Yes, probably | Yes | Yes (if cleavage faster than fibrillisation at high TL conc.) | Yes | Yes | Yes | Yes |
Applications
Label-free drug screening
Thermofluor has been extensively used in drug screening campaigns.[28][29][30][31][32][33][34][35][36] Since thermofluor basically detects high affinity binding sites for small molecules on proteins, it can find hits that bind to active site subsites, cofactor sites, or allosteric binding sites with equal efficacy.The method typically requires the use of screening compound concentrations at >10x the desired binding threshold. Setting 5 micromolar as a reasonable hit threshold consequently requires a test ligand concentration of 50 to 100micromolar in the sample well. For most drug compound libraries, where many compounds are not soluble beyond ~100 micromolar, screening multiple compounds is consequently not feasible owing to solubility issues. Thermoflour screens do not require the development of custom screening reagents (e.g. cleavable substrate analogs), do not require any radioactive reagents, and are generally less sensitive to the effects of compounds that are chemically reactive with protein active site residues, and that consequently show up as undesirable hits in enzyme activity screens.
Drug lead optimization
Thermofluor measurements of Tm can be quantitatively related to drug Kd values,[37] although this requires the additional calorimetric measurements of the target proteins’ enthalpy of unfolding, determined using DSC. The dynamic range of the thermofluor assay is very large, so that the same assay can be used to find micromolar hits and to optimize sub-nanomolar leads, making the method particularly useful in the development of QSAR relationships for lead optimization.
Studies of enzyme mechanism
Many proteins require the simultaneous or sequential binding of multiple substrates, cofactors, and/or allosteric effectors. Thermofluor studies of molecules that bind to active site subsites, cofactor sites, or allosteric binding sites can help elucidate specific features of enzyme mechanism that can be important in the design of effective drug screening campaigns.[38]
Protein stabilization for optimized isolation
Thermofluor pre-screens can be performed that sample a wide range of pH, ionic strength and additives such as added metal ions and cofactors. The generation of a protein response surface is useful for establishing optimal assay conditions and can frequently led to improved purification scheme as required to support HTS campaigns and biophysical studies.[39][40]
Characterization of engineered proteins
Many applications of protein engineering for drug discovery or biophysics applications involve modification of the protein amino acid sequence through truncation, domain fusions, site-specific modifications or random mutagenesis. Thermofluor provides a high throughput method for the evaluation of the effects of such sequence variations on protein stability as well as means for developing stabilizing conditions if required.[41][42]
Optimization of protein crystallization conditions
Although proteins are dynamic structures in solution, formation of protein crystals is expected to be favored when all molecules lie in their lowest energy conformation. Thermofluor evaluation of conditions that stabilize proteins is consequently a useful strategy for finding optimal crystallization conditions [43][44][45]
Screening for Inhibitors of Protein-Protein Interactions of Modulators of Protein Conformational Changes
Since Thermofluor is a label-free assay that detects small molecule binding to high affinity binding sites on a target protein, it is well suited to finding small molecule inhibitors of protein-protein interactions or allosteric modulation sites.[46][47] Of course, whether or not a protein-protein interaction is ultimately “druggable” with a small molecule requires the presence of a suitable binding site on the target protein that provides enough local energetic interactions to allow specific drug binding.
ThermoFluor of Membrane Proteins
Membrane proteins are often isolated in the presence of hydrophobic solubilizing agents that can partition hydrophobic-binding dyes like 1,8ANS and SYPRO orange and generate a fluorescence background that obscures observation of a Thermofluor protein melting signal. Nevertheless, careful optimization of conditions (e.g. to avoid micelle formation of the solubilizing agent) can often produce satisfactory assay conditions [48][49]
Decrypting Proteins of Unknown Biological Function
The biochemical function of protein targets identified through gene knockout or proteomics approaches are often obscure if they have low amino acid sequence homology with proteins of known function. In many cases some useful information can be gained through the identification of binding cofactors or substrate analogs in classifying protein function, information useful in using Thermofluor can assist in “decrypting” the function of proteins whose biochemical function might otherwise be unknown.[50] [51]
Parallel Thermal Shift Assays
Recent developments have extended thermal shift approaches to the analysis of ligand interactions in complex mixtures, including intact cells. Initial observations of individual proteins using fast parallel proteolysis (FastPP) showed that stabilization by ligand binding could impart resistance to proteolytic digestion with thermolysin. Protection relative to reference was quantified through either protein staining on gels or Western blotting with a labeling antibody directed to a tag fused to the target protein.[52] CETSA, for cellular thermal shift assay, is a method that monitors the stabilization effect of drug binding through the prevention of irreversible protein precipitation, which is usually initiated when a protein becomes thermally denatured. In CETSA, aliquots of cell lysate are transiently heated to different temperatures, following which samples are centrifuged to separate soluble fractions from precipitated proteins. The presence of the target protein in each soluble fraction is determined by Western blotting and used to construct a CESTA melt curve that can inform regarding in vivo targeting, drug distribution, and bioavailability.[53] Both FastPP and CETSA generally require antibodies to facilitate target detection, and consequently are generally used in contexts where the target identity is known a priori. Newer developments seek to merge aspects of FastPP and CETSA approaches, by assessing the ligand-dependent dependent proteolytic protection of targets in cells using mass spectroscopy (MS) to detect shifts in proteolysis patterns associated with protein stabilization.[54] Present implementations still require a priori knowledge of expected targets to facilitate data analysis, but improvements in MS data collection strategies, together with the use of improved computational tools and database structures can potentially allow the approach to be used for de novo target decryption on the total cell proteome scale. This would be a major advance for drug discovery since it would allow the identification of discrete molecular targets (as well as off-target interactions) for drugs identified through high-content cellular or phenotypic drug screens.
References
- ↑ Koshland, DE (February 1958). "Application of a Theory of Enzyme Specificity to Protein Synthesis.". Proceedings of the National Academy of Sciences of the United States of America. 44 (2): 98–104. doi:10.1073/pnas.44.2.98. PMC 335371
 . PMID 16590179.
. PMID 16590179. - ↑ Linderstrøm-Lang, K.; Schellman, J. A. (1959). "Protein structure and enzyme activity". The Enzymes. 1 (2): 443–510.
- ↑ Waldron, KJ; Rutherford, JC; Ford, D; Robinson, NJ (13 August 2009). "Metalloproteins and metal sensing.". Nature. 460 (7257): 823–30. doi:10.1038/nature08300. PMID 19675642.
- ↑ Saiki, RK; Gelfand, DH; Stoffel, S; Scharf, SJ; Higuchi, R; Horn, GT; Mullis, KB; Erlich, HA (29 January 1988). "Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase.". Science. 239 (4839): 487–91. doi:10.1126/science.2448875. PMID 2448875.
- ↑ Dupeux, F; Röwer, M; Seroul, G; Blot, D; Márquez, JA (November 2011). "A thermal stability assay can help to estimate the crystallization likelihood of biological samples.". Acta Crystallographica Section D. 67 (Pt 11): 915–9. doi:10.1107/s0907444911036225. PMID 22101817.
- ↑ Ericsson, UB; Hallberg, BM; Detitta, GT; Dekker, N; Nordlund, P (15 October 2006). "Thermofluor-based high-throughput stability optimization of proteins for structural studies.". Analytical Biochemistry. 357 (2): 289–98. doi:10.1016/j.ab.2006.07.027. PMID 16962548.
- ↑ Grøftehauge, MK; Hajizadeh, NR; Swann, MJ; Pohl, E (1 January 2015). "Protein-ligand interactions investigated by thermal shift assays (TSA) and dual polarization interferometry (DPI).". Acta Crystallographica Section D. 71 (Pt 1): 36–44. doi:10.1107/s1399004714016617. PMID 25615858.
- ↑ Semisotnov, GV; Rodionova, NA; Razgulyaev, OI; Uversky, VN; Gripas', AF; Gilmanshin, RI (January 1991). "Study of the "molten globule" intermediate state in protein folding by a hydrophobic fluorescent probe.". Biopolymers. 31 (1): 119–28. doi:10.1002/bip.360310111. PMID 2025683.
- ↑ Pantoliano, MW; Petrella, EC; Kwasnoski, JD; Lobanov, VS; Myslik, J; Graf, E; Carver, T; Asel, E; Springer, BA; Lane, P; Salemme, FR (December 2001). "High-density miniaturized thermal shift assays as a general strategy for drug discovery.". Journal of Biomolecular Screening. 6 (6): 429–40. doi:10.1177/108705710100600609. PMID 11788061.
- ↑ Lo, MC; Aulabaugh, A; Jin, G; Cowling, R; Bard, J; Malamas, M; Ellestad, G (1 September 2004). "Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery.". Analytical Biochemistry. 332 (1): 153–9. doi:10.1016/j.ab.2004.04.031. PMID 15301960.
- ↑ Niesen, FH; Berglund, H; Vedadi, M (2007). "The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability.". Nature protocols. 2 (9): 2212–21. doi:10.1038/nprot.2007.321. PMID 17853878.
- ↑ Kohlstaedt, M; von der Hocht, I; Hilbers, F; Thielmann, Y; Michel, H (May 2015). "Development of a Thermofluor assay for stability determination of membrane proteins using the Na(+)/H(+) antiporter NhaA and cytochrome c oxidase.". Acta Crystallographica Section D. 71 (Pt 5): 1112–22. doi:10.1107/s1399004715004058. PMID 25945577.
- ↑ Ciulli, A; Abell, C (December 2007). "Fragment-based approaches to enzyme inhibition.". Current opinion in biotechnology. 18 (6): 489–96. doi:10.1016/j.copbio.2007.09.003. PMID 17959370.
- ↑ Kranz, JK; Schalk-Hihi, C (2011). "Protein thermal shifts to identify low molecular weight fragments.". Methods in enzymology. 493: 277–98. doi:10.1016/B978-0-12-381274-2.00011-X. PMID 21371595.
- ↑ Alexandrov, AI; Mileni, M; Chien, EY; Hanson, MA; Stevens, RC (March 2008). "Microscale fluorescent thermal stability assay for membrane proteins.". Structure (London, England : 1993). 16 (3): 351–9. doi:10.1016/j.str.2008.02.004. PMID 18334210.
- ↑ Menzen, T; Friess, W (February 2013). "High-throughput melting-temperature analysis of a monoclonal antibody by differential scanning fluorimetry in the presence of surfactants.". Journal of pharmaceutical sciences. 102 (2): 415–28. doi:10.1002/jps.23405. PMID 23212746.
- ↑ Minde, DP; Maurice, MM; Rüdiger, SG (2012). "Determining biophysical protein stability in lysates by a fast proteolysis assay, FASTpp.". PLOS ONE. 7 (10): e46147. doi:10.1371/journal.pone.0046147. PMC 3463568. PMID 23056252.
- ↑ Jafari, R; Almqvist, H; Axelsson, H; Ignatushchenko, M; Lundbäck, T; Nordlund, P; Martinez Molina, D (September 2014). "The cellular thermal shift assay for evaluating drug target interactions in cells.". Nature protocols. 9 (9): 2100–22. doi:10.1038/nprot.2014.138. PMID 25101824.
- ↑ Kreisig, T; Prasse, AA; Zscharnack, K; Volke, D; Zuchner, T (8 July 2014). "His-tag protein monitoring by a fast mix-and-measure immunoassay.". Scientific Reports. 4: 5613. doi:10.1038/srep05613. PMID 25000910.
- ↑ Ashok, Y; Nanekar, R; Jaakola, VP (December 2015). "Defining thermostability of membrane proteins by western blotting.". Protein engineering, design & selection : PEDS. 28 (12): 539–42. doi:10.1093/protein/gzv049. PMID 26384510.
- ↑ "ThermoFAD, a Thermofluor-adapted flavin ad hoc detection system for protein folding and ligand binding". FEBS J. 276: 2833–40. 2009. doi:10.1111/j.1742-4658.2009.07006.x. PMID 19459938.
- ↑ Mancusso, R; Karpowich, NK; Czyzewski, BK; Wang, DN (December 2011). "Simple screening method for improving membrane protein thermostability.". Methods (San Diego, Calif.). 55 (4): 324–9. doi:10.1016/j.ymeth.2011.07.008. PMC 3220791. PMID 21840396.
- ↑ Czyzewski, BK; Wang, DN (11 March 2012). "Identification and characterization of a bacterial hydrosulphide ion channel.". Nature. 483 (7390): 494–7. doi:10.1038/nature10881. PMID 22407320.
- ↑ Mancusso, R; Gregorio, GG; Liu, Q; Wang, DN (22 November 2012). "Structure and mechanism of a bacterial sodium-dependent dicarboxylate transporter.". Nature. 491 (7425): 622–6. doi:10.1038/nature11542. PMID 23086149.
- ↑ Kawate, T; Gouaux, E (April 2006). "Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins.". Structure (London, England : 1993). 14 (4): 673–81. doi:10.1016/j.str.2006.01.013. PMID 16615909.
- ↑ Hattori, M; Hibbs, RE; Gouaux, E (8 August 2012). "A fluorescence-detection size-exclusion chromatography-based thermostability assay for membrane protein precrystallization screening.". Structure (London, England : 1993). 20 (8): 1293–9. doi:10.1016/j.str.2012.06.009. PMID 22884106.
- ↑ Lebon, G; Bennett, K; Jazayeri, A; Tate, CG (10 June 2011). "Thermostabilisation of an agonist-bound conformation of the human adenosine A(2A) receptor.". Journal of Molecular Biology. 409 (3): 298–310. doi:10.1016/j.jmb.2011.03.075. PMID 21501622.
- ↑ Pantoliano, MW; Petrella, EC; Kwasnoski, JD; Lobanov, VS; Myslik, J; Graf, E; Carver, T; Asel, E; Springer, BA; Lane, P; Salemme, FR (December 2001). "High-density miniaturized thermal shift assays as a general strategy for drug discovery.". Journal of Biomolecular Screening. 6 (6): 429–40. doi:10.1177/108705710100600609. PMID 11788061.
- ↑ Ciulli, A; Abell, C (December 2007). "Fragment-based approaches to enzyme inhibition.". Current opinion in biotechnology. 18 (6): 489–96. doi:10.1016/j.copbio.2007.09.003. PMID 17959370.
- ↑ Kranz, JK; Schalk-Hihi, C (2011). "Protein thermal shifts to identify low molecular weight fragments.". Methods in enzymology. 493: 277–98. doi:10.1016/B978-0-12-381274-2.00011-X. PMID 21371595.
- ↑ Alexandrov, AI; Mileni, M; Chien, EY; Hanson, MA; Stevens, RC (March 2008). "Microscale fluorescent thermal stability assay for membrane proteins.". Structure (London, England : 1993). 16 (3): 351–9. doi:10.1016/j.str.2008.02.004. PMID 18334210.
- ↑ Lo, MC; Aulabaugh, A; Jin, G; Cowling, R; Bard, J (2004). "Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery.". Analytical Biochemistry. 332: 153–159.
- ↑ DeSantis, KA; Reinking, JL (2016). "Use of Differential Scanning Fluorimetry to Identify Nuclear Receptor Ligands.". Methods Molecular Biology. 1443: 21–30.
- ↑ Bergsdorf, CI; Otti, J (2010). "Affinity-based screening techniques: their impact and benefit to increase the number of high quality leads.". Expert Opinion in Drug Discovery). 5: 1095–1107.
- ↑ Cummings, MD; Farnum, MA; Nelen, MI (2006). "Universal screening methods and applications of ThermoFluor.". Journal of Biomolecular Screening. 11: 854–863.
- ↑ Groftehauge, MK; Hajizadeh, NR; Swann, MJ; Pohl, E (January 2015). "Protein-ligand interactions investigated by thermal shift assays (TSA) and dual polarization interferometry (DPI.". Acta Cryst. Section D Biological Crystallography). 71: 36–44. doi:10.1107/s1399004714016617. PMID 25615858.
- ↑ Matulis, D; Kranz, JK; Salemme, FR; Todd, MJ (2005). "Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor.". Biochemistry. 44: 5258–5266.
- ↑ Lea, WA; Simeonov, A (April 2012). "Differential scanning fluorometry signatures as indicators of enzyme inhibitor mode of action: case study of glutathione S-transferase.". PLoS One. 7 (4): e366219. doi:10.1371/journal.pone.0036219.
- ↑ Niesen, FH; Berglund, H; Vedadi, M (2007). "The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability.". Nature Protocols. 2 (9): 2212–2221. doi:10.1038/nprot.2007.321. PMID 17853878.
- ↑ Mezzasalama, TM (April 2007). "Enhancing recombinant protein quality and yield by protein stability profiling.". Journal of Biomolecular Screening. 12 (3): 418–428. PMID 17438070.
- ↑ Nettleship, JE; Brown, J; Groves, MR; Geerlof, A (2008). "Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering.". Methods Molecular Biology. 426: 299–318. doi:10.1007/978-1-60327-058-8_19. PMID 18542872.
- ↑ lavinder, JJ; Hari, SB; Sullivan, BJ; Magliery, TJ (2009). "High-throughput thermal scanning: a general, rapid dye-binding thermal shift screen for protein engineering.". Journal of The American Chemical Society. 131: 3794–3795.
- ↑ Ericsson, UB; Halberg, BM; Detitta, GT; Dekker, N; Nordlund, P (October 2006). "Thermofluor-based high-throughput stability optimization of proteins for structural studies.". Analytical Biochemistry. 357 (2): 289–298. doi:10.1016/j.ab.2006.07.027. PMID 16962548.
- ↑ Vedadi, M; Niesen, FH; Allali-Hassani, A; Fedorov, OY; Finerty, PJ Jr (2006). "Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination.". Proceedings of the National Academy of Science USA. 103: 15835–15840.
- ↑ Dupeux, F; Rower, M; Seroul, G; Blot, D; Marquez, JA (November 2011). "A thermal stability assay can help to estimate the crystallization likelihood of biological samples.". Acta Cryst Section D Biological Crystallography. 67: 915–919. doi:10.1107/s0907444911036225. PMID 22101817.
- ↑ Grasberger, BL (February 2005). "Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells.". Journal of Medicinal Chemistry. 48 (4): 909–912. PMID 15715460.
- ↑ Charveriat, M (2009). "New inhibitors of prion replication that target the amyloid precursor.". Journal Gen Virology. 90: 1294–1301. PMID 19264641.
- ↑ Kohlstaedt, M; von der Hocht, I; Hilbers, F; Thielmann, Y; Michel, H (May 2005). "Development of a Thermofluor assay for stability determination of membrane proteins using the Na(+)/H(+) antiporter NhaA and cytochrome c oxidase.". Acta Cryst. Section D. Biological crystallography. 71: 1112–1122. doi:10.1107/s1399004715004058. PMID 25945577.
- ↑ Alexandrov, AI; Mileni, M; Chien, EY; Hanson, MA; Stevens, RC (March 2008). "Microscale fluorescent thermal stability assay for membrane proteins.". Structure (London, England : 1993). 16 (3): 351–9. doi:10.1016/j.str.2008.02.004. PMID 18334210.
- ↑ Todd, MJ; Cummings, MD; Nelen, MI (2005). "Affinity assays for decrypting protein targets of unknown function.". Drug Discovery Today. Technologies. 2: 267–273.
- ↑ Carver, TE; Bordeau, B; Cummings, MD; Petrella, EC; Pucci, MJ; Zawadzke, LE; Dougherty, BA; Tredupl, JA; Bryson, JW; Yanchunas, J Jr.; Doyle, ML; Witmer, MR; Nelen, MI; Desjarlais, RL; Jaeger, EPL; Devine, H; Asel, ED; Springer, BA; Bone, R; Salemme, FR; Todd, M (2005). "Decrypting the Biochemical Function of an Essential Gene from Streptococcus pneumoniae using Thermofluor Technology.". Journal of Biological Chemistry. 280: 11704–11712.
- ↑ Minde, DP; Maurice, MM; Rüdiger, SG (2012). "Determining biophysical protein stability in lysates by a fast proteolysis assay, FASTpp.". PLOS ONE. 7 (10): e46147. doi:10.1371/journal.pone.0046147. PMC 3463568. PMID 23056252.
- ↑ Martinez, MD; Jafari, R; Ignatushchenko, M; Seki, T; Larsson, EA; Dan, C; Sreekumar, L; Cao, Y; Nordlund, P (July 2013). "Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay.". Science. 341 (6141): 84–87. doi:10.1126/science.1233606.
- ↑ Savitski, MM; Reinhard, FB; Franken, H; Werner, T; Savitski, MF; Eberhard, D; Martinez, MD; Jafari, R; Dovega, RB; Klaeger, S; Kuster, B; Nordlund, P; Bantscheff, M; Drewes, G (October 2014). "Tracking cancer drugs in living cells by thermal profiling of the proteome.". Science. 346 (6205): 125–128.