Cahn–Ingold–Prelog priority rules
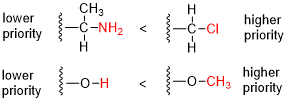
The Cahn–Ingold–Prelog (CIP) sequence rules, named for organic chemists R.S. Cahn, C.K. Ingold, and V. Prelog—alternatively termed the CIP priority rules, system, or conventions—are a standard process used in organic chemistry to completely and unequivocally name a stereoisomer of a molecule.[1] [2]:26 The purpose of the CIP system is to assign an R or S descriptor to each stereocenter and an E or Z descriptor to each double bond so that the configuration of the entire molecule can be specified uniquely by including the descriptors in its systematic name. A molecule may contain any number of stereocenters and any number of double bonds, and each usually gives rise to two possible isomers. (A molecule with an integer describing the number of its stereogenic centers will usually have stereoisomers, diastereomers each having an associated pair of enantiomers.[3][4] The CIP sequence rules contribute to the precise naming of every stereoisomer of every organic and organometallic molecule with all atoms of ligancy of fewer than 4 (but including ligancy of 6 as well, this term referring to the "number of neighboring atoms" bonded to a center).[2]:26f[4]



The key article setting out the CIP sequence rules was published in 1966,[5] and was followed by further refinements,[6][7] before it was incorporated into the rules of the International Union of Pure and Applied Chemistry, the official body that defines organic nomenclature.[2]:26ff The IUPAC presentation of the rules constitute the official, formal standard for their use, and it notes that "the method has been developed to cover all compounds with ligancy up to 4... and… [extended to the case of] ligancy 6… [as well as] for all configurations and conformations of such compounds."[2]:26ff Nevertheless, though the IUPAC documentation presents a thorough introduction, it includes the caution that "it is essential to study the original papers, especially the 1966 paper, before using the sequence rule for other than fairly simple cases."[2]:26f
Steps for naming
The steps for naming molecules using the CIP system are often presented as:
- Identification of stereocenters and double bonds;
- Assignment of priorities to the groups attached to each stereocenter or double-bonded atom; and
- Assignment of R/S and E/Z descriptors.
Assignment of priorities
R/S and E/Z descriptors are assigned by using a system for ranking priority of the groups attached to each stereocenter. This procedure, often known as the sequence rules, is the heart of the CIP system.
- Compare the atomic number (Z) of the atoms directly attached to the stereocenter; the group having the atom of higher atomic number receives higher priority.
- If there is a tie, we must consider the atoms at distance 2 from the stereocenter—as a list is made for each group of the atoms bonded to the one directly attached to the stereocenter. Each list is arranged in order of decreasing atomic number. Then the lists are compared atom by atom; at the earliest difference, the group containing the atom of higher atomic number receives higher priority.
- If there is still a tie, each atom in each of the two lists is replaced with a sub-list of the other atoms bonded to it (at distance 3 from the stereocenter), the sub-lists are arranged in decreasing order of atomic number, and the entire structure is again compared atom by atom. This process is repeated, each time with atoms one bond farther from the stereocenter, until the tie is broken.
Isotopes
If two groups differ only in isotopes, atomic masses are used at each step to break ties in atomic number.
Double and triple bonds

If an atom A is double-bonded to an atom B, A is treated as being singly bonded to two atoms: B and a ghost atom that is a duplicate of B (has the same atomic number) but is not attached to anything except A. When B is replaced with a list of attached atoms, A itself is excluded in accordance with the general principle of not doubling back along a bond that has just been followed. A triple bond is handled the same way except that A and B both have duplicated 'ghost' atoms.[2]:28
Geometric Isomers
If two substituents on an atom are geometric isomers, the Z-isomer has higher priority than the E-isomer.
Cycles
To handle a molecule containing one or more cycles, one must first expand it into a tree (called a hierarchical digraph by the authors) by traversing bonds in all possible paths starting at the stereocenter. When the traversal encounters an atom through which the current path has already passed, a ghost atom is generated in order to keep the tree finite. A single atom of the original molecule may appear in many places (some as ghosts, some not) in the tree.
Assigning descriptors
Stereocenters: R/S
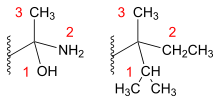
After the substituents of a stereocenter have been assigned their priorities, the molecule is oriented in space so that the group with the lowest priority is pointed away from the observer. If the substituents are numbered from 1 (highest priority) to 4 (lowest priority), then the sense of rotation of a curve passing through 1, 2 and 3 distinguishes the stereoisomers. A center with a clockwise sense of rotation is an R or rectus center and a center with a counterclockwise sense of rotation is an S or sinister center. The names are derived from the Latin for right and left, respectively.[8]
-1%2C2%2C3-trichlorocyclopentane.svg.png)
A practical method of determining whether an enantiomer is R or S is by using the right-hand rule: one wraps the molecule with the fingers in the direction 1→2→3. If the thumb points in the direction of the 4th substituent, the enantiomer is R. Otherwise, it's S.
It is possible in rare cases that two substituents on an atom differ only in their absolute configuration (R or S). If the relative priorities of these substituents need to be established, R takes priority over S. When this happens, the descriptor of the stereocenter is a lowercase letter (r or s) instead of the uppercase letter normally used.[9]
Double bonds: E/Z
For alkenes and similar double bonded molecules, the same prioritizing process is followed for the substituents. In this case, it is the placing of the two highest priority substituents with respect to the double bond which matters. If both high priority substituents are on the same side of the double bond, i.e. in the cis configuration, then the stereoisomer is assigned a Z or Zusammen configuration. If, by contrast they are in a trans configuration, then the stereoisomer is assigned an E or Entgegen configuration. In this case the identifying letters are derived from German for 'together' and 'in opposition to', respectively.
Examples
The following are examples of application of the nomenclature.
| R/S assignments for several compounds | |
|---|---|
 |
The hypothetical molecule bromochlorofluoroiodomethane shown in its R-configuration would be a very simple chiral compound. The priorities are assigned based on atomic number (Z): iodine (Z = 53) > bromine (Z = 35) > chlorine (Z = 17) > fluorine (Z = 9). Allowing fluorine (lowest priority) to point away from the viewer the rotation is clockwise hence the R-assignment. |
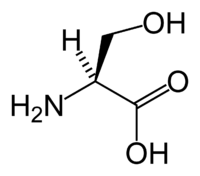 |
In the assignment of L-serine highest priority is given to the nitrogen atom (Z = 7) in the amino group (NH2). Both the methylalcohol group (CH2OH ) and the carboxylic acid group (COOH) have carbon atoms (Z = 6) but priority is given to the latter because the carbon atom in the COOH group is connected to a second oxygen (Z=8) whereas in the CH2OH group carbon is connected to a hydrogen atom (Z=1). Lowest priority is given to the hydrogen atom and as this atom points away from the viewer the counterclockwise decrease in priority over the three remaining substituents completes the assignment as S. |
-Carvone.svg.png) |
The stereocenter in S-carvone is connected to one hydrogen atom (not shown, priority 4) and three carbon atoms. The isopropene group has priority 1 (carbon atoms only) and for the two remaining carbon atoms priority is decided with the carbon atoms two bonds removed from the stereocenter, one part of the keto group (O,O,C priority 2) and one part of an alkene (H,C,C priority 3). The resulting counterclockwise rotation results in a S. |
Describing multiple centers
If a compound has more than one stereocenter each center is denoted by either R or S. For example, ephedrine exists with both (1R,2S) and (1S,2R) configuration, known as enantiomers. This compound also exists with a (1R,2R) and (1S,2S) configuration. The last two stereoisomers are not ephedrine, but pseudoephedrine. All isomers are 2-methylamino-1-phenyl-1-propanol in systematic nomenclature. Pseudoephedrine is chemically distinct from ephedrine with only the three-dimensional configuration in space, as notated by the Cahn–Ingold–Prelog rules. The two compounds, ephedrine and pseudoephedrine, are diastereomers, or stereoisomers that are not enantiomers. They have different names because, as diastereomers, they have different chemical properties.
In pairs of enantiomers, all descriptors are opposite: R,R and S,S or R,S and S,R. Diastereomers have one descriptor in common: R,S and R,R or S,R and S,S. This holds true for compounds with more than two stereocenters; if at least one descriptor is the same in both pairs, the compounds are diastereomers. If all the stereocenters are opposite, they are enantiomers.
Relative configuration
The relative configuration of two stereoisomers may be denoted by the descriptors R and S with an asterisk (*). "R*,R*" means two centers having identical configurations (R,R or S,S); "R*,S*" means two centers having opposite configurations (R,S or S,R). To begin, the lowest numbered (according to IUPAC systematic numbering) stereogenic center is given the R* descriptor.
To designate two anomers the relative stereodescriptors alpha (α) and beta (β) are used. In the α anomer the anomeric carbon and the reference atom do have opposite configurations (R,S or S,R), whereas in the β anomer they are the same (both R or both S). [10]
Faces

Stereochemistry also plays a role assigning faces to trigonal molecules such as ketones. A nucleophile in a nucleophilic addition can approach the carbonyl group from two opposite sides or faces. When an achiral nucleophile attacks acetone, both faces are identical and there is only one reaction product. When the nucleophile attacks butanone, the faces are not identical (enantiotopic) and a racemic product results. When the nucleophile is a chiral molecule diastereoisomers are formed. When one face of a molecule is shielded by substituents or geometric constraints compared to the other face the faces are called diastereotopic. The same rules that determine the stereochemistry of a stereocenter (R or S) also apply when assigning the face of a molecular group. The faces are then called the re-faces and si-faces. In the example displayed on the right, the compound acetophenone is viewed from the re face. Hydride addition as in a reduction process from this side will form the S-enantiomer and attack from the opposite Si face will give the R-enantiomer. However, one should note that adding a chemical group to the prochiral center from the re-face will not always lead to an S stereocenter, as the priority of the chemical group has to be taken into account. That is, the absolute stereochemistry of the product is determined on its own and not by considering which face it was attacked from. In the above-mentioned example, if chloride (Cl-) was added to the prochiral center from the re-face, this would result in an R-enantiomer.
References
- ↑ March, J. Advanced Organic Chemistry (3rd ed.). ISBN 0-471-85472-7.
- 1 2 3 4 5 6 Cross, L.C; Klyne, W. (1974). Rules for the Nomenclature of Organic Chemistry: Section E: Stereochemistry (Recommendations 1974) (PDF). ISBN 978-0-08-021019-3. Archived from the original (PDF) on 2016-04-07.
- ↑ Clayden, Jonathan; Greeves, Nick & Warren, Stuart (2012). Organic Chemistry (2nd ed.). Oxford, UK: Oxford University Press. pp. 316f. ISBN 0199270295. Retrieved 2 February 2016.
- 1 2 The "usually" has its basis in the fact that molecules with chiral centers nevertheless may have mirror planes of symmetry, e.g. meso compounds, that make some of the stereoisomers "degenerate" (identical), so that this mathematical expression overestimates the number. See Clayden, op. cit., p. 317.
- ↑ Cahn, R.S.; Ingold, C.K.; Prelog, V. (1966). "Specification of Molecular Chirality". Angewandte Chemie International Edition. 5 (4): 385–415. doi:10.1002/anie.196603851.
- ↑ E.g., see Prelog, V. & Helmchen, G. (1982). "Basic Principles of the CIP-System and Proposals for a Revision". Angewandte Chemie International Edition. 21 (8): 567–58. doi:10.1002/anie.198205671.
- ↑ These two papers together define the bulk of the CIP system; the papers provide a number of additional rules beyond the main points covered above, including describing less common forms of stereoisomerism (such as chiral axes and planes), and resolving more difficult priority assignments.
- ↑ Klein, David R. (2013-12-31). Organic Chemistry (2nd ed.). Wiley. p. 203. ISBN 978-1118454312.
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "pseudo-asymmetric carbon atom".
- ↑ IUPAC Goldbook relative configuration