Hajdu–Cheney syndrome
| Hajdu–Cheney syndrome | |
|---|---|
 | |
| Hajdu-Cheney | |
| Classification and external resources | |
| OMIM | 102500 |
| DiseasesDB | 31486 |
| MeSH | D031845 |
Hajdu–Cheney syndrome, also called acroosteolysis with osteoporosis and changes in skull and mandible, arthrodentoosteodysplasia, and Cheney syndrome,[1] is an extremely rare autosomal dominant congenital disorder[2][3] of the connective tissue characterized by severe and excessive bone resorption leading to osteoporosis and a wide range of other possible symptoms. Mutations in the NOTCH2 gene, identified in 2011, cause HCS. HCS is so rare that only about 70 cases have been reported worldwide, since the discovery of the syndrome in 1948.
Signs and symptoms
Hajdu–Cheney syndrome causes many issues with an individual’s connective tissues. Some general characteristics of an individual with Hajdu–Cheney syndrome include bone flexibility and deformities, short stature, delayed acquisition of speech and motor skills, dolichocephalic skull, Wormian bone, small maxilla, hypoplastic frontal sinuses, basilar impression, joint laxity, bulbous finger tips, and severe osteoporosis. Wormian bone occurs when extra bones appear between cranial sutures. Fetuses with Hajdu–Cheney syndrome often will not be seen to unclench their hands on obstetrical ultrasound. They may also have low-set ears and their eyes may be farther apart than on a usual child, called hypertelorism. Children's heads can have some deformities in their shape and size (plagiocephaly). Early tooth loss and bone deformities, such as serpentine tibiae and fibulae, are also common in those affected.
Genetics
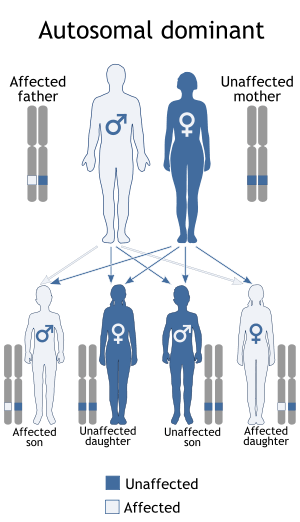
Hajdu–Cheney syndrome is a monogenic disorder. The disorder is inherited and controlled by a single pair of genes. A single copy of the mutant gene on an autosome causes HCS. HCS is an autosomal dominant disorder, only one parent with the defective gene is needed to pass the disorder to the offspring. Mutations within the last coding exon of NOTCH2 that remove the PEST domain and escape the nonsense-mediated mRNA decay have been shown to be the main cause of Hajdu–Cheney syndrome.[4][5][6] The NOTCH2 gene plays a very important role in skeletogenesis. Mutations of NOTCH2 that seem to cause HCS occur in the last coding exon of the gene (exon 34). These mutations remove PEST domains, which mediate proteosomal destruction of the protein. These PEST domains are removed due to the premature stop codon in the amino acid sequence. All HCS alleles are observed to have premature protein destruction before the PEST sequence is fully translated. The result is a mature NOTCH2 gene with a partially completed PEST sequence. In some cases, no PEST sequence at all is seen. This leads to the no proteosomal destruction of the protein. The NOTCH2 gene is ubiquitously expressed in all embryonic tissue. When researching HCS in mice, the homozygous deletion of NOTCH2 leads to death. This observation is important because it explains how the HCS phenotype is not isolated to only one system of the body. NOTCH2 is also shown to regulate RANK-L osteoclastogenesis, which is the production of functional osteoclasts. Osteoclasts are the component that breaks bone down. This is why bone loss is observed in HCS patients, due to the overactivation of RANK-L.
Pathogenesis
The mechanism thought to cause HCS is an abnormality in osteoblast and osteoid function. These are major components of bone development, and the low function of each leads to the weak bones that characterize HCS.
Diagnosis
One of the main methods of pinpointing a NOTCH2 mutation that leads to HCS is through whole genome sequencing. This is then followed by exome capture by means of in-solution hybridization. The exome part of the genome consists of exons. Parallel sequencing follows the hybridization, which results in about 3.5 Gb of sequence data. These sequence data are then analyzed. Through sequence analysis and symptom presentation in HCS patients, this proves to be the most definitive method of diagnosis.
Types
Laboratory testing reveals multiple mutations of HCS. Two genetic variants result in sporadic HCS symptoms, which are HCS-02 and HCS-03. These mutations produce symptoms that come and go, but have been present de novo. HCS-03 was identified as the variant that is passed through afflicted family members and presents symptoms throughout the lifetime of the individual. All variants of HCS lead to the same premature termination of PEST sequences which compromise normal function of NOTCH2. NOTCH has four different receptors, which have an affinity for similar ligands. They are classified as single-pass transmembrane receptors.
Treatment
Since about 2002, some patients with this disorder have been offered drug therapy with bisphosphonates (a class of osteoporosis drugs) to treat problems with bone resorption associated with the bone breakdown and skeletal malformations that characterize this disorder. Brand names include Actonel (risedronate/alendronate), made by Merck Pharmaceuticals. Other drugs include Pamidronate, made by Novartis and Strontium Ranelate, made by Eli Lilly. However, for more progressive cases, surgery and bone grafting are necessary.
Eponym
It is named after Nicholas Hajdu (1908-1987), a Hungarian-English radiologist working in the UK and William D. Cheney, MD (1899-1985), a US radiologist.
References
- ↑ Online Mendelian Inheritance in Man (OMIM) 102500
- ↑ Crifasi, P. A.; Patterson, M. C.; Bonde, D.; Michels, V. V. (Jun 1997). "Severe Hajdu-Cheney syndrome with upper airway obstruction". American Journal of Medical Genetics. 70 (3): 261–266. doi:10.1002/(SICI)1096-8628(19970613)70:3<261::AID-AJMG9>3.0.CO;2-Z. PMID 9188663.
- ↑ Brennan AM, Pauli RM (May 2001). "Hajdu--Cheney syndrome: evolution of phenotype and clinical problems". Am. J. Med. Genet. 100 (4): 292–310. doi:10.1002/1096-8628(20010515)100:4<292::AID-AJMG1308>3.0.CO;2-4. PMID 11343321.
- ↑ A Simpson, Michael; Irving, Melita D; Asilmaz, Esra; Gray, Mary J; Dafou, Dimitra; Elmslie, Frances V; Mansour, Sahar; Holder, Sue E; Brain, Caroline E (2011). "Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss". Nature Genetics. 43 (4): 303–305. doi:10.1038/ng.779. PMID 21378985. Retrieved 2011-03-07.
- ↑ Isidor, Bertrand; Pierre Lindenbaum (2011). "Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis". Nature Genetics. 43 (4): 306–308. doi:10.1038/ng.778. PMID 21378989.
- ↑ Majewski, J; Schwartzentruber, Jeremy A.; Caqueret, Aurore; Patry, Lysanne; Marcadier, Janet; Fryns, Jean-Pierre; Boycott, Kym M.; Ste-Marie, Louis-Georges; McKiernan, Fergus E. (2011). "Mutations in NOTCH2 in families with Hajdu-Cheney syndrome". Hum Mutat. 32: n/a–n/a. doi:10.1002/humu.21546. PMID 21681853.
Further reading
- Ades L. C., Morris L. L., Haan E. A. (1993). "Hydrocephalus in Hajdu–Cheney syndrome". Journal of Medical Genetics. 30 (2): 175. doi:10.1136/jmg.30.2.175.
- Bamshad M. J., Ng S. B., Bigham A. W., Tabor H. K., Emond M. J., Nickerson D. A., Shendure J. (2011). "Exome sequencing as a tool for Mendelian disease gene discovery". Nature Reviews Genetics. 12 (11): 745–755. doi:10.1038/nrg3031.
- Brennan A. M., Pauli R. M. (2001). "Hajdu‐Cheney syndrome: Evolution of phenotype and clinical problems". American Journal of Medical Genetics. 100 (4): 292–310. doi:10.1002/1096-8628(20010515)100:4<292::AID-AJMG1308>3.0.CO;2-4. PMID 11343321.
- Cremin Bryan, Spranger Jürgen, Beighton M. D. Peter (1982). "Wormian bones in osteogenesis imperfecta and other disorders". Skeletal radiology. 8 (1): 35–38. doi:10.1007/BF00361366.
- Iwaya T., Taniguchi K., Watanabe J., Iinuma K., Hamazaki Y., Yoshikawa S. (1979). "Hajdu–Cheney syndrome". Archives of orthopaedic and traumatic surgery. 95 (4): 293–302. doi:10.1007/bf00389701.