Pancreatic neuroendocrine tumor
| Pancreatic neuroendocrine tumor | |
|---|---|
 | |
| Classification and external resources | |
| Specialty | Oncology |
| ICD-10 | C25.4 |
| eMedicine | med/ |
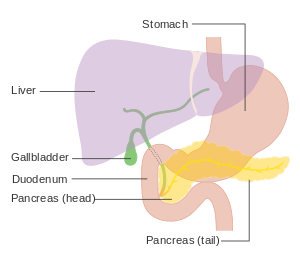
Pancreatic neuroendocrine tumors (PanNETs, PETs, or PNETs), often referred to as "islet cell tumors",[1][2] or "pancreatic endocrine tumors"[3][4] are neuroendocrine neoplasms that arise from cells of the endocrine (hormonal) and nervous system within the pancreas.
PanNETs are a type of neuroendocrine tumor, representing about one third of gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Many PanNETs are benign, while some are malignant. Aggressive PanNET tumors have traditionally been termed "islet cell carcinoma".
PanNETs are quite distinct from the usual form of pancreatic cancer, the majority of which are adenocarcinomas, which arises in the exocrine pancreas. Only 1 or 2% of clinically significant pancreas neoplasms are PanNETs.
Types
PanNETs are sometimes abbreviated as PETs or PNETs: such use should not to be confused with the primitive neuroectodermal tumor (PNET).
The majority of PanNETs are benign, while some are malignant. The World Health Organization (WHO) classification scheme places neuroendocrine tumors into three main categories, which emphasize the tumor grade rather than the anatomical origin.[3] In practice, those tumors termed well or intermediately differentiated PanNETs in the WHO scheme are sometimes called "islet cell tumors." The high grade subtype, termed neuroendocrine cancer (NEC) in the WHO scheme, is synonymous with "islet cell carcinoma".
Signs and symptoms
Some PanNETs do not cause any symptoms, in which case they may be discovered incidentally on a CT scan performed for a different purpose.[5]:43–44 Symptoms such as abdominal or back pain or pressure, diarrhea, indigestion, or yellowing of the skin and whites of the eyes can arise from the effects of a larger PanNET tumor, either locally or at a metastasis.[6] About 40% of PanNETS have symptoms related to excessive secretion of hormones or active polypeptides and are accordingly labeled as "functional"; the symptoms reflect the type of hormone secreted, as discussed below. Up to 60% of PanNETs are nonsecretory or nonfunctional, in which there is no secretion, or the quantity or type of products, such as pancreatic polypeptide (PPoma), chromogranin A, and neurotensin, do not cause a clinical syndrome although blood levels may be elevated.[7] In total, 85% of PanNETs have an elevated blood marker.[2]
Functional tumors are often classified by the hormone most strongly secreted, for example:
- gastrinoma: the excessive gastrin causes Zollinger–Ellison syndrome (ZES) with peptic ulcers and diarrhea
- insulinoma:[8] hypoglycemia occurs with concurrent elevations of insulin, proinsulin and C peptide[9]
- glucagonoma: the symptoms are not all due to glucagon elevations,[9] and include a rash, sore mouth, altered bowel habits, venous thrombosis, and high blood glucose levels[9]
- VIPoma, producing excessive vasoactive intestinal peptide, which may cause profound chronic watery diarrhea and resultant dehydration, hypokalemia, and achlorhydria (WDHA or pancreatic cholera syndrome)
- somatostatinoma: these rare tumors are associated with elevated blood glucose levels, achlorhydria, cholelithiasis, and diarrhea[9]
- less common types include ACTHoma, CRHoma, calcitoninoma, GHRHoma, GRFoma, and parathyroid hormone–related peptide tumor
In these various types of functional tumors, the frequency of malignancy and the survival prognosis have been estimated dissimilarly, but a pertinent accessible summary is available.[10]
Diagnosis
Imaging
- CT, MRI, EUS, SRS[11]
- Octreotide scaning
MEN1
Personal and family history should be evaluated for MEN1.
Staging
The 2010 WHO classification of tumors of the digestive system grades all the neuroendocrine tumors into three categories, based on their degree of cellular differentiation (from well-differentiated "NET G1" through to poorly-differentiated "NET G3"). The NCCN recommends use of the same AJCC-UICC staging system as pancreatic adenocarcinoma.[5]:52 Using this scheme, the stage by stage outcomes for PanNETs are dissimilar to pancreatic exocrine cancers.[12] A different TNM system for PanNETs has been proposed by The European Neuroendocrine Tumor Society.[13]
- Pancreatic neuroendocrine tumor staging (AJCC)
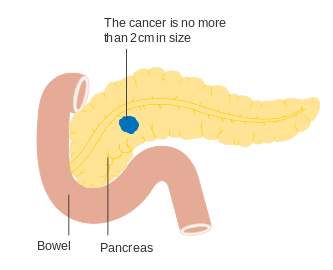 Stage T1
Stage T1 Stage T2
Stage T2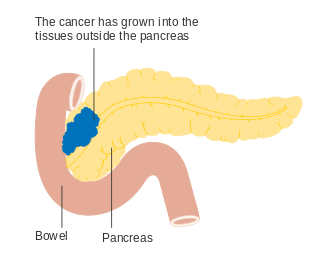 Stage T3
Stage T3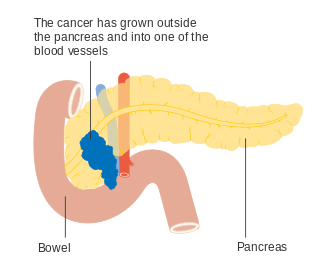 Stage T4
Stage T4_CRUK_178.svg.png) Involvement of nearby lymph nodes – Stage N1
Involvement of nearby lymph nodes – Stage N1_CRUK_179.svg.png) Metastasis – stage M1
Metastasis – stage M1
Treatment
In general, treatment for PanNET encompasses the same array of options as other neuroendocrine tumors, as discussed in that main article. However, there are some specific differences, which are discussed here.[5]
In functioning PanNETs, octreotide is usually recommended prior to biopsy[5]:21 or surgery[5]:45 but is generally avoided in insulinomas to avoid profound hypoglycemia.[5]:69
PanNETs in MEN1 are often multiple, and thus require different treatment and surveillance strategies.[5]
Some PanNETs are more responsive to chemotherapy than are gastroenteric carcinoid tumors. Several agents have shown activity.[9] In well differentiated PanNETs, chemotherapy is generally reserved for when there are no other treatment options. Combinations of several medicines have been used, such as doxorubicin with streptozocin and fluorouracil (5-FU)[9][14] and capecitabine with temozolomide.[14] Although marginally effective in well-differentiated PETs, cisplatin with etoposide has some activity in poorly differentiated neuroendocrine cancers (PDNECs),[9] particularly if the PDNEC has an extremely high Ki-67 score of over 50%.[5]:30
Several targeted therapy agents have been approved in PanNETs by the FDA based on improved progression-free survival (PFS):
- everolimus (Afinitor) is labeled for treatment of progressive neuroendocrine tumors of pancreatic origin in patients with unresectable, locally advanced or metastatic disease.[15][16] The safety and effectiveness of everolimus in carcinoid tumors have not been established.[15][16]
- sunitinib (Sutent) is labeled for treatment of progressive, well-differentiated pancreatic neuroendocrine tumors in patients with unresectable locally advanced or metastatic disease.[17][18] Sutent also has approval from the European Commission for the treatment of 'unresectable or metastatic, well-differentiated pancreatic neuroendocrine tumors with disease progression in adults'.[19] A phase III study of sunitinib treatment in well differentiated pNET that had worsened within the past 12 months (either advanced or metastatic disease) showed that sunitinib treatment improved progression-free survival (11.4 months vs. 5.5 months), overall survival, and the objective response rate (9.3% vs. 0.0%) when compared with placebo.[20]
Genetics
DNA mutation analysis in well-differentiated pancreatic neuroendocrine tumors identified four important findings:[21][22]
- as expected, the genes mutated in NETs, MEN1, ATRX, DAXX, TSC2, PTEN and PIK3CA,[21] are different from the mutated genes previously found in pancreatic adenocarcinoma.[23][24]
- one in six well-differentiated pancreatic NETs have mutations in mTOR pathway genes, such as TSC2, PTEN and PIK3CA.[21] The sequencing discovery might allow selection of which NETs would benefit from mTOR inhibition such as with everolimus, but this awaits validation in a clinical trial.
- mutations affecting a new cancer pathway involving ATRX and DAXX genes were found in about 40% of pancreatic NETs.[21] The proteins encoded by ATRX and DAXX participate in chromatin remodeling of telomeres;[25] these mutations are associated with a telomerase-independent maintenance mechanism termed ALT (alternative lengthening of telomeres) that results in abnormally long telomeric ends of chromosomes.[25]
- ATRX/DAXX and MEN1 mutations were associated with a better prognosis.[21]
References
- ↑ Burns WR, Edil BH (March 2012). "Neuroendocrine pancreatic tumors: guidelines for management and update". Current treatment options in oncology. 13 (1): 24–34. doi:10.1007/s11864-011-0172-2. PMID 22198808.
- 1 2 Pancreatic Neuroendocrine Tumors (Islet Cell Tumors) Treatment (PDQ) Health Professional Version. National Cancer Institute. March 7, 2014.
- 1 2 The PanNET denomination is in line with current WHO guidelines. Historically, PanNETs have also been referred to by a variety of terms, and are still often called "islet cell tumors" or "pancreatic endocrine tumors". See: Klimstra DS, Modlin IR, Coppola D, et al. (August 2010). "The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems" (PDF). Pancreas. 39 (6): 707–12. doi:10.1097/MPA.0b013e3181ec124e. PMID 20664470.
- ↑ Oberg, K (2010). "Pancreatic endocrine tumors". Seminars in Oncology. 37 (6): 594–618. doi:10.1053/j.seminoncol.2010.10.014. PMID 21167379.
- 1 2 3 4 5 6 7 8 "Neuroendocrine tumors, NCCN Guidelines Version 1.2015" (PDF). NCCN Guidelines. National Comprehensive Cancer Network, Inc. November 11, 2014. Retrieved December 25, 2014.
- ↑ Pancreatic Neuroendocrine Tumors (Islet Cell Tumors) Treatment (PDQ®) National Cancer Institute
- ↑ Jensen RT, Berna MJ, Bingham DB, Norton JA (2008). "Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies". Cancer. 113 (7 Suppl): 1807–1843. doi:10.1002/cncr.23648. PMC 2574000
 . PMID 18798544.
. PMID 18798544. - ↑ Grant C (2005). "Insulinoma". Best Practice & Research Clinical Gastroenterology. 19 (5): 783–798. doi:10.1016/j.bpg.2005.05.008. PMID 16253900.
- 1 2 3 4 5 6 7 Benson AB, Myerson RJ, and Sasson AR. Pancreatic, neuroendocrine GI, and adrenal cancers. Cancer Management: A Multidisciplinary Approach 13th edition 2010. ISBN 978-0-615-41824-7 Text is available electronically (but may require free registration) at http://www.cancernetwork.com/cancer-management/pancreatic/article/10165/1802606
- ↑ Ramage JK, Davies AH, Ardill J, et al. (Jun 2005). "Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours". Gut. 54. Suppl 4 (suppl_4): iv1–16. doi:10.1136/gut.2004.053314. PMC 1867801. PMID 15888809.
- ↑ Ong, ES Neoplasms of the Endocrine Pancreas Workup - Imaging Studies
- ↑ National Cancer Institute. Pancreatic Neuroendocrine Tumors (Islet Cell Tumors) Treatment (PDQ®) Incidence and Mortality
- ↑ Öberg K, Knigge U, Kwekkeboom D, Perren A (October 2012). "Neuroendocrine gastro-entero-pancreatic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up". Annals of Oncology. 23 Suppl 7: vii124–30. doi:10.1093/annonc/mds295. PMID 22997445. (Table 5 outlines the proposed TNM staging system for PanNETs.)
- 1 2 Tejani MA, Saif MW (2014). "Pancreatic neuroendocrine tumors: does chemotherapy work?". JOP. 15 (2): 132–4. PMID 24618436.
- 1 2 Everolimus Approved for Pancreatic Neuroendocrine Tumors. The ASCO Post. May 15, 2011, Volume 2, Issue 8 http://ascopost.com/articles/may-15-2011/everolimus-approved-for-pancreatic-neuroendocrine-tumors/
- 1 2 http://www.pharma.us.novartis.com/product/pi/pdf/afinitor.pdf
- ↑ National Cancer Institute. Cancer Drug Information. FDA Approval for Sunitinib Malate. Pancreatic Neuroendocrine Tumors http://www.cancer.gov/cancertopics/druginfo/fda-sunitinib-malate
- ↑ http://labeling.pfizer.com/ShowLabeling.aspx?id=607
- ↑ "Pfizer Scores New Approval for Sutent in Europe". 2 Dec 2010.
- ↑ Raymond E, Dahan L, Raoul JL, et al. (2011). "Sunitinib malate for the treatment of pancreatic neuroendocrine tumors". N Engl J Med. 364 (6): 501–13. doi:10.1056/NEJMoa1003825. PMID 21306237.
- 1 2 3 4 5 Jiao, Y.; Shi, C.; Edil, B. H.; De Wilde, R. F.; Klimstra, D. S.; Maitra, A.; Schulick, R. D.; Tang, L. H.; Wolfgang, C. L.; Choti, M. A.; Velculescu, V. E.; Diaz Jr, L. A.; Vogelstein, B.; Kinzler, K. W.; Hruban, R. H.; Papadopoulos, N. (2011). "DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors". Science. 331 (6021): 1199–1203. doi:10.1126/science.1200609. PMC 3144496. PMID 21252315.
- ↑ McKenna, L. R.; Edil, B. H. (2014). "Update on pancreatic neuroendocrine tumors". Gland surgery. 3 (4): 258–275. doi:10.3978/j.issn.2227-684X.2014.06.03. PMC 4244504. PMID 25493258.
- ↑ Jones, S.; Zhang, X.; Parsons, D. W.; Lin, J. C. -H.; Leary, R. J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; Hong, S. -M.; Fu, B.; Lin, M. -T.; Calhoun, E. S.; Kamiyama, M.; Walter, K.; Nikolskaya, T.; Nikolsky, Y.; Hartigan, J.; Smith, D. R.; Hidalgo, M.; Leach, S. D.; Klein, A. P.; Jaffee, E. M.; Goggins, M.; Maitra, A.; Iacobuzio-Donahue, C.; Eshleman, J. R.; Kern, S. E.; Hruban, R. H. (2008). "Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses". Science. 321 (5897): 1801–1806. doi:10.1126/science.1164368. PMC 2848990. PMID 18772397.
- ↑ Harada, T.; Chelala, C.; Crnogorac-Jurcevic, T.; Lemoine, N. R. (2009). "Genome-Wide Analysis of Pancreatic Cancer Using Microarray-Based Techniques". Pancreatology. 9 (1–2): 13–24. doi:10.1159/000178871. PMID 19077451.
- 1 2 Heaphy CM, De Wilde RF, Jiao Y, et al. (2011). "Altered Telomeres in Tumors with ATRX and DAXX Mutations". Science. 333 (6041): 425. doi:10.1126/science.1207313. PMC 3174141. PMID 21719641.