Telomerase
| RNA-directed DNA polymerase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
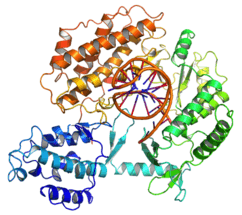
A conceptual diagram showing the protein component of telomerase (TERT) in grey and the RNA component (TR) in yellow | |||||||||
| Identifiers | |||||||||
| EC number | 2.7.7.49 | ||||||||
| CAS number | 9068-38-6 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / EGO | ||||||||
| |||||||||

Telomerase, also called terminal transferase,[1] is a ribonucleoprotein that adds a species-dependent telomere repeat sequence to the 3' end of telomeres. A telomere is a region of repetitive sequences at each end of a eukaryotic chromosomes in most eukaryotes. Telomeres protect the end of the chromosome from DNA damage or from fusion with neighbouring chromosomes. The fruit fly Drosophila melanogaster lacks telomerase, but instead uses retrotransposons to maintain telomeres.[2]
Telomerase is a reverse transcriptase enzyme that carries its own RNA molecule (e.g., with the sequence "CCCAAUCCC" in vertebrates) which is used as a template when it elongates telomeres. Telomerase, active in normal stem cells and most cancer cells, is normally absent from, or at very low levels in, most somatic cells.
History
The existence of a compensatory mechanism for telomere shortening was first found by Soviet biologist Alexey Olovnikov in 1973,[3] who also suggested the telomere hypothesis of aging and the telomere's connections to cancer.
Telomerase in the ciliate Tetrahymena was discovered by Carol W. Greider and Elizabeth Blackburn in 1984.[4] Together with Jack W. Szostak, Greider and Blackburn were awarded the 2009 Nobel Prize in Physiology or Medicine for their discovery.[5]
The role of telomeres and telomerase in cell aging and cancer was established by scientists at biotechnology company Geron with the cloning of the RNA and catalytic components of human telomerase[6] and the development of a polymerase chain reaction (PCR) based assay for telomerase activity called the TRAP assay, which surveys telomerase activity in multiple types of cancer.[7]
Human telomerase structure
The molecular composition of the human telomerase enzyme complex was determined by Dr Scott Cohen and his team at the Children's Medical Research Institute (Sydney Australia) and consists of two molecules each of human telomerase reverse transcriptase (TERT), telomerase RNA (TR or TERC), and dyskerin (DKC1).[8] The genes of telomerase subunits, which include TERT,[9] TERC,[10] DKC1[11] and TEP1,[12] are located on different chromosomes. The human TERT gene (hTERT) is translated into a protein of 1132 amino acids.[13] TERT polypeptide folds with (and carries) TERC, a non-coding RNA (451 nucleotides long). TERT has a 'mitten' structure that allows it to wrap around the chromosome to add single-stranded telomere repeats.
TERT is a reverse transcriptase, which is a class of enzyme that creates single-stranded DNA using single-stranded RNA as a template.
The protein consists of four conserved domains (RNA-Binding Domain (TRBD), fingers, palm and thumb), organized into a ring configuration that shares common features with retroviral reverse transcriptases, viral RNA polymerases and bacteriophage B-family DNA polymerases.[14][15]
TERT proteins from many eukaryotes have been sequenced.[16]
Function
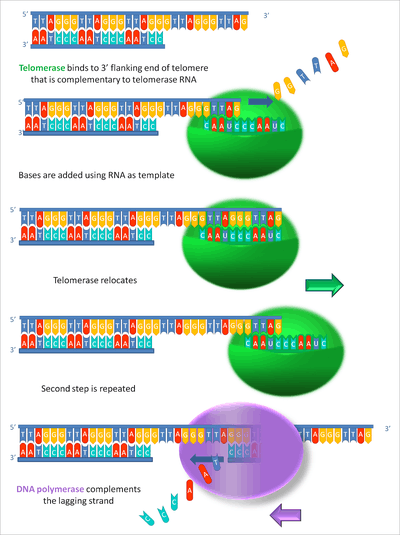
By using TERC, TERT can add a six-nucleotide repeating sequence, 5'-TTAGGG (in vertebrates, the sequence differs in other organisms) to the 3' strand of chromosomes. These TTAGGG repeats (with their various protein binding partners) are called telomeres. The template region of TERC is 3'-CAAUCCCAAUC-5'.[17]
Telomerase can bind the first few nucleotides of the template to the last telomere sequence on the chromosome, add a new telomere repeat (5'-GGTTAG-3') sequence, let go, realign the new 3'-end of telomere to the template, and repeat the process. Telomerase reverses telomere shortening.
Clinical implications
Aging
Telomerase replaces short bits of DNA known as telomeres, which are otherwise shortened when a cell divides via mitosis.
In normal circumstances, absent telomerase, if a cell divides recursively, at some point the progeny reach their Hayflick limit,[18] which is believed to be between 50–70 cell divisions. At the limit the cells become senescent and cell division stops.[19] Telomerase allows each offspring to replace the lost bit of DNA allowing the cell line to divide without ever reaching the limit. This same unbounded growth is a feature of cancerous growth.[20]
Embryonic stem cells express telomerase, which allows them to divide repeatedly and form the individual. In adults, telomerase is highly expressed only in cells that need to divide regularly especially in male sperm cells but also in epidermal cells,[21] in activated T cell[22] and B cell[23] lymphocytes, as well as in certain adult stem cells, but in the great majority of cases somatic cells do not express telomerase.[24]
A comparative biology study of mammalian telomeres indicated that telomere length of some mammalian species correlates inversely, rather than directly, with lifespan, and concluded that the contribution of telomere length to lifespan is unresolved.[25] Telomere shortening does not occur with age in some postmitotic tissues, such as in the rat brain.[26] In humans, skeletal muscle telomere lengths remain stable from ages 23 –74.[27] In baboon skeletal muscle, which consists of fully differentiated post-mitotic cells, less than 3% of myonuclei contain damaged telomeres and this percentage does not increase with age.[28] Thus, telomere shortening does not appear to be a major factor in the aging of the differentiated cells of brain or skeletal muscle. In human liver, cholangiocytes and hepatocytes show no age-related telomere shortening.[29] Another study found little evidence that, in humans, telomere length is a significant biomarker of normal aging with respect to important cognitive and physical abilities.[30]
Some experiments have raised questions on whether telomerase can be used as an anti-aging therapy, namely, the fact that mice with elevated levels of telomerase have higher cancer incidence and hence do not live longer. Telomerase also favors tumorogenesis, which leads to questions about its potential as an anti-aging therapy.[31] On the other hand, one study showed that activating telomerase in cancer-resistant mice by overexpressing its catalytic subunit extended lifespan.[32]
Exposure of T lymphocytes from HIV-infected human donors to a small molecule telomerase activator (TAT2) retards telomere shortening, increases proliferative potential and enhances cytokine/chemokine production and antiviral activity.[33]
A study that focused on Ashkenazi Jews found that long-lived subjects inherited a hyperactive version of telomerase.[34]
Mice engineered to block the gene that produces telomerase, unless they are given a certain drug, aged at a much faster rate, and died at about six months, instead of reaching the average mouse lifespan, about three years. Administering the drug at 6 months turned on telomerase production and caused their organs to be "rejuvenated," restored fertility, and normalized their ability to detect or process odors.[35][36]
A 2012 study reported that introducing the TERT gene into healthy one-year-old mice using an engineered adeno-associated virus led to a 24% increase in lifespan, without any increase in cancer.[37]
Premature aging
Premature aging syndromes including Werner syndrome, Ataxia telangiectasia, Ataxia-telangiectasia like disorder, Bloom syndrome, Fanconi anemia and Nijmegen breakage syndrome are associated with short telomeres.[38] However, the genes that have mutated in these diseases all have roles in the repair of DNA damage and the increased DNA damage may, itself, be a factor in the premature aging (see DNA damage theory of aging). An additional role in maintaining telomere length is an active area of investigation.
Cancer
In vitro, when cells approach the Hayflick limit, the time to senescence can be extended by inactivating the tumor suppressor proteins - p53 and Retinoblastoma protein (pRb). Cells that have been so-altered eventually undergo an event termed a "crisis" when the majority of the cells in the culture die. Sometimes, a cell does not stop dividing once it reaches crisis. In a typical situation, the telomeres are shortened[39] and chromosomal integrity declines with every subsequent cell division. Exposed chromosome ends are interpreted as double-stranded breaks (DSB) in DNA; such damage is usually repaired by reattaching (religating) the broken ends together. When the cell does this due to telomere-shortening, the ends of different chromosomes can be attached to each other. This solves the problem of lacking telomeres, but during cell division anaphase, the fused chromosomes are randomly ripped apart, causing many mutations and chromosomal abnormalities. As this process continues, the cell's genome becomes unstable. Eventually, either fatal damage is done to the cell's chromosomes (killing it via apoptosis), or an additional mutation that activates telomerase occurs.
With telomerase activation some types of cells and their offspring become immortal (bypass the Hayflick limit), thus avoiding cell death as long as the conditions for their duplication are met. Many cancer cells are considered 'immortal' because telomerase activity allows them to live much longer than any other somatic cell, which, combined with uncontrollable cell proliferation[40] is why they can form tumors. A good example of immortal cancer cells is HeLa cells, which have been used in laboratories as a model cell line since 1951.
While this method of modeling human cancer in cell culture is effective and has been used for many years by scientists, it is also very imprecise. The exact changes that allow for the formation of the tumorigenic clones in the above-described experiment are not clear. Scientists addressed this question by the serial introduction of multiple mutations present in a variety of human cancers. This has led to the identification of mutation combinations that form tumorigenic cells in a variety of cell types. While the combination varies by cell type, the following alterations are required in all cases: TERT activation, loss of p53 pathway function, loss of pRb pathway function, activation of the Ras or myc proto-oncogenes, and aberration of the PP2A protein phosphatase. That is to say, the cell has an activated telomerase, eliminating the process of death by chromosome instability or loss, absence of apoptosis-induction pathways, and continued mitosis activation.
This model of cancer in cell culture accurately describes the role of telomerase in actual human tumors. Telomerase activation has been observed in ~90% of all human tumors,[41] suggesting that the immortality conferred by telomerase plays a key role in cancer development. Of the tumors without TERT activation,[42] most employ a separate pathway to maintain telomere length termed Alternative Lengthening of Telomeres (ALT ).[43] The exact mechanism behind telomere maintenance in the ALT pathway is unclear, but likely involves multiple recombination events at the telomere.
Elizabeth Blackburn et al., identified the upregulation of 70 genes known or suspected in cancer growth and spread through the body, and the activation of glycolysis, which enables cancer cells to rapidly use sugar to facilitate their programmed growth rate (roughly the growth rate of a fetus).[44]
Approaches to controlling telomerase and telomeres for cancer therapy include gene therapy, immunotherapy, small-molecule and signal pathway inhibitors.[45]
Drugs
The ability to maintain functional telomeres may be one mechanism that allows cancer cells to grow in vitro for decades.[46] Telomerase activity is necessary to preserve many cancer types and is inactive in somatic cells, creating the possibility that telomerase inhibition could selectively repress cancer cell growth with minimal side effects.[47] If a drug can inhibit telomerase in cancer cells, the telomeres of successive generations will progressively shorten, limiting tumor growth.[48]
Telomerase is a good biomarker for cancer detection because most human cancers cells express high levels of it. Telomerase activity can be identified by its catalytic protein domain (hTERT). This is the rate-limiting step in telomerase activity. It is associated with many cancer types. Various cancer cells and fibroblasts transformed with hTERT cDNA have high telomerase activity, while somatic cells do not. Cells testing positive for hTERT have positive nuclear signals. Epithelial stem cell tissue and its early daughter cells are the only noncancerous cells in which hTERT can be detected. Since hTERT expression is dependent only on the number of tumor cells within a sample, the amount of hTERT indicates the severity of a cancer.[49]
The expression of hTERT can also be used to distinguish benign tumors from malignant tumors. Malignant tumors have higher hTERT expression than benign tumors. Real-time reverse transcription polymerase chain reaction (RT-PCR) quantifying hTERT expression in various tumor samples verified this varying expression.[50]
The lack of telomerase does not affect cell growth, until the telomeres are short enough to cause cells to “die or undergo growth arrest”. However, inhibiting telomerase alone is not enough to destroy large tumors. It must be combined with surgery, radiation, chemotherapy or immunotherapy.[49]
Cells may reduce their telomere length by only 50-252 base pairs per cell division, which can lead to a long lag phase.[51][52]
Immunotherapy
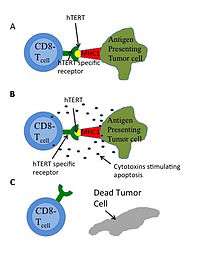
Immunotherapy successfully treats some kinds of cancer, such as melanoma. This treatment involves manipulating a human’s immune system to destroy cancerous cells. Humans have two major antigen identifying lymphocytes: CD8+ cytotoxic T-lymphocytes (CTL) and CD4+ helper T-lymphocytes that can destroy cells. Antigen receptors on CTL can bind to a 9-10 amino acid chain that is presented by the major histocompatibility complex (MHC) as in Figure 4. HTERT is a potential target antigen. Immunotargeting should result in relatively few side effects since hTERT expression is associated only with telomerase and is not essential in almost all somatic cells.[53] GV1001 uses this pathway.[45] Experimental drug and vaccine therapies targeting active telomerase have been tested in mouse models, and clinical trials have begun.
In 2014 Geron Corporation received permission to resume a trial of its drug imetelstat for myelofibrosis after addressing FDA concerns over liver toxicity.[54][55] Geron licensee Merck had approval of an IND for one vaccine type. Imetelstat (GRN163L) binds directly to the telomerase's RNA template. One 2015 study reported that Imetelstat caused partial or complete remission in seven of 33 patients, while a second reported that it decreased blood platelet levels in all 18 study patients with essential thrombocythemia, a disorder in which the body overproduces blood platelets, increasing the risk of blood clots.[56]
Most of the harmful cancer-related effects of telomerase are dependent on an intact RNA template. Cancer stem cells that use an alternative method of telomere maintenance are still killed when telomerase's RNA template is blocked or damaged.
Telomerase Vaccines
Two telomerase vaccines have been developed: GRNVAC1 and GV1001. GRNVAC1 isolates dendritic cells and the RNA that codes for the telomerase protein and puts them back into the patient to make cytotoxic T cells that kill the telomerase-active cells. GV1001 is a peptide from the active site of hTERT and is recognized by the immune system that reacts by killing the telomerase-active cells.[45]
Targeted apoptosis
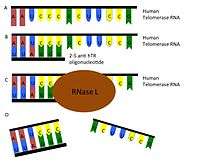
Another independent approach is to use oligoadenylated anti-telomerase antisense oligonucleotides and ribozymes to target telomerase RNA, inducing dissociation and apoptosis (Figure 5). The fast induction of apoptosis through antisense binding may be a good alternative to the slower telomere shortening.[51]
Heart disease, diabetes and quality of life
Blackburn also discovered that mothers caring for very sick children have shorter telomeres when they report that their emotional stress is at a maximum and that telomerase was active at the site of blockages in coronary artery tissue, possibly accelerating heart attacks.
In 2009, it was shown that the amount of telomerase activity significantly increased following psychological stress. Across the sample of patients telomerase activity in increased peripheral blood mononuclear cells by 18% one hour after the end of the stress.[57]
E. V. Gostjeva et al. found no differences between colon cancer stem cells and fetal colon stem cells.[58]
A study in 2010 found that there was "significantly greater" telomerase activity in participants than controls after a three-month meditation retreat.[59]
Telomerase deficiency has been linked to diabetes mellitus and impaired insulin secretion in mice, due to loss of pancreatic insulin-producing cells.[60]
Rare human diseases
Mutations in TERT have been implicated in predisposing patients to aplastic anemia, a disorder in which the bone marrow fails to produce blood cells, in 2005.[61]
Cri du chat syndrome (CdCS) is a complex disorder involving the loss of the distal portion of the short arm of chromosome 5. TERT is located in the deleted region, and loss of one copy of TERT has been suggested as a cause or contributing factor of this disease.[62]
Dyskeratosis congenita (DC) is a disease of the bone marrow that can be caused by some mutations in the telomerase subunits.[63] In the DC cases, about 35% cases are X-linked-recessive on the DKC1 locus[64] and 5% cases are autosomal dominant on the TERT[65] and TERC[66] loci.
Patients with DC have severe bone marrow failure manifesting as abnormal skin pigmentation, leucoplakia (a white thickening of the oral mucosa) and nail dystrophy, as well as a variety of other symptoms. Individuals with either TERC or DKC1 mutations have shorter telomeres and defective telomerase activity in vitro versus other individuals of the same age.[67]
In one family autosomal dominant DC was linked to a heterozygous TERT mutation.[68] These patients also exhibited an increased rate of telomere-shortening, and genetic anticipation (i.e., the DC phenotype worsened with each generation).
See also
References
- ↑ What are telomeres and telomerase?
- ↑ Pardue ML, DeBaryshe PG (2011). "Retrotransposons that maintain chromosome ends" (PDF). PNAS. 108 (51): 20317–20324. doi:10.1073/pnas.1100278108. PMC 3251079
 . PMID 21821789.
. PMID 21821789. - ↑ Olovnikov AM (September 1973). "A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon". J. Theor. Biol. 41 (1): 181–90. doi:10.1016/0022-5193(73)90198-7. PMID 4754905.
- ↑ Greider CW, Blackburn EH (December 1985). "Identification of a specific telomere terminal transferase activity in Tetrahymena extracts". Cell. 43 (2 Pt 1): 405–13. doi:10.1016/0092-8674(85)90170-9. PMID 3907856.
- ↑ "The Nobel Prize in Physiology or Medicine 2009". The Nobel Foundation. 2009-10-05. Retrieved 2010-10-23.
- ↑ Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC, Yu J (September 1995). "The RNA component of human telomerase". Science. 269 (5228): 1236–41. doi:10.1126/science.7544491. PMID 7544491.
- ↑ Kim, N.; Piatyszek, M.; Prowse, K.; Harley, C.; West, M.; Ho, P.; Coviello, G.; Wright, W.; Weinrich, S. (1994). "Specific association of human telomerase activity with immortal cells and cancer". Science. 266 (5193): 2011–5. doi:10.1126/science.7605428. PMID 7605428.
- ↑ Cohen S, Graham M, Lovrecz G, Bache N, Robinson P, Reddel R (2007). "Protein composition of catalytically active human telomerase from immortal cells". Science. 315 (5820): 1850–3. doi:10.1126/science.1138596. PMID 17395830.
- ↑ "HGNC database of human gene names - HUGO Gene Nomenclature Committee". genenames.org.
- ↑ HGNC - TERC
- ↑ HGNC - DKC1
- ↑ HGNC - TEP1
- ↑ NCBI - telomerase reverse transcriptase isoform 1
- ↑ Gillis AJ, Schuller AP, Skordalakes E. Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature. 2008 Oct 2;455(7213):633-7
- ↑ Mitchell M, Gillis A, Futahashi M, Fujiwara H, Skordalakes E. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat Struct Mol Biol. 2010 Apr;17(4):513-8
- ↑ NCBI - telomerase reverse transcriptase
- ↑ Gavory G, Farrow M, Balasubramanian S (October 2002). "Minimum length requirement of the alignment domain of human telomerase RNA to sustain catalytic activity in vitro". Nucleic Acids Res. 30 (20): 4470–80. doi:10.1093/nar/gkf575. PMC 137139. PMID 12384594.
- ↑ Hayflick L, Moorhead PS (1961). "The serial cultivation of human diploid cell strains". Exp Cell Res. 25 (3): 585–621. doi:10.1016/0014-4827(61)90192-6. PMID 13905658.
- ↑ Siegel, L (2013). Are Telomeres the Key to Aging and Cancer? The University of Utah. Retrieved 30 September 2013
- ↑ Hanahan D, Weinberg RA (March 2011). "Hallmarks of cancer: the next generation". Cell. 144 (5): 646–74. doi:10.1016/j.cell.2011.02.013. PMID 21376230.
- ↑ Härle-Bachor C, Boukamp P (1996). "Telomerase activity in the regenerative basal layer of the epidermis inhuman skin and in immortal and carcinoma-derived skin keratinocytes". PNAS. 93 (13): 6476–6481. PMC 39048. PMID 8692840.
- ↑ Barsov EV (2011). "Telomerase and primary T cells: biology and immortalization for adoptive immunotherapy". Immunotherapy (journal). 3 (3): 407–421. doi:10.2217/imt.10.107. PMC 3120014. PMID 21395382.
- ↑ Bougel S, Renaud S, Braunschweig R, Loukinov D, Morse HC 3rd, Bosman FT, Lobanenkov V, Benhattar J (2010). "PAX5 activates the transcription of the human telomerase reverse transcriptase gene in B cells". The Journal of Pathology. 220 (1): 87–96. doi:10.1002/path.2620. PMC 3422366. PMID 19806612.
- ↑ Cong YS (2002). "Human Telomerase and Its Regulation". Microbiology and Molecular Biology Reviews. 66 (3): 407–425. doi:10.1128/MMBR.66.3.407-425.2002. PMC 120798. PMID 12208997.
- ↑ Gomes, NM; Ryder, OA; Houck, ML; Charter, SJ; Walker, W,; Forsyth, NR; Austad, SN; Venditti, C; Pagel, M; Shay, JW; Wright, WE (2011). "Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination.". Aging Cell. 105 (5): 761–768. doi:10.1111/j.1474-9726.2011.00718.x. PMC 3387546. PMID 21518243.
- ↑ Cherif H, Tarry JL, Ozanne SE, Hales CN (2003). "Ageing and telomeres: a study into organ- and gender-specific telomere shortening". Nucleic Acids Res. 31 (5): 1576–1583. doi:10.1093/nar/gkg208. PMID 12595567.
- ↑ "Regenerative potential of human skeletal muscle during aging.". Aging Cell. 1 (2): 132–9. Dec 2002. doi:10.1046/j.1474-9728.2002.00017.x. PMID 12882343.
- ↑ Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U (2007). "Accumulation of senescent cells in mitotic tissue of aging primates". Mech Ageing Dev. 128 (1): 36–44. doi:10.1016/j.mad.2006.11.008. PMID 17116315.
- ↑ Verma S, Tachtatzis P, Penrhyn-Lowe S, Scarpini C, Jurk D, Von Zglinicki T, Coleman N, Alexander GJ (2012). "Sustained telomere length in hepatocytes and cholangiocytes with increasing age in normal liver". Hepatology. 56 (4): 1510–1520. doi:10.1002/hep.25787. PMID 22504828.
- ↑ "Telomere length and aging biomarkers in 70-year-olds: the Lothian Birth Cohort 1936.". Neurobiol Aging. 33 (7): 1486.e3–8. Jul 2012. doi:10.1016/j.neurobiolaging.2010.11.013. PMID 21194798.
- ↑ de Magalhães JP, Toussaint O (2004). "Telomeres and telomerase: a modern fountain of youth?". Rejuvenation Res. 7 (2): 126–33. doi:10.1089/1549168041553044. PMID 15312299.
- ↑ Tomás-Loba A, Flores I, Fernández-Marcos PJ, Cayuela ML, Maraver A, Tejera A, Borrás C, Matheu A, Klatt P, Flores JM, Viña J, Serrano M, Blasco MA (November 2008). "Telomerase reverse transcriptase delays aging in cancer-resistant mice". Cell. 135 (4): 609–22. doi:10.1016/j.cell.2008.09.034. PMID 19013273.
- ↑ Fauce SR, Jamieson BD, Chin AC, Mitsuyasu RT, Parish ST, Ng HL, Kitchen CM, Yang OO, Harley CB, Effros RB (November 2008). "Telomerase-Based Pharmacologic Enhancement of Antiviral Function> of Human CD8+ T Lymphocytes". J. Immunol. 181 (10): 7400–6. doi:10.4049/jimmunol.181.10.7400. PMC 2682219. PMID 18981163.
- ↑ Atzmon G, Cho M, Cawthon RM, Budagov T, Katz M, Yang X, Siegel G, Bergman A, Huffman DM, Schechter CB, Wright WE, Shay JW, Barzilai N, Govindaraju DR, Suh Y (January 2010). "Genetic variation in human telomerase is associated with telomere length in Ashkenazi centenarians". Proc. Natl. Acad. Sci. U.S.A. 107 Suppl 1 (suppl_1): 1710–7. doi:10.1073/pnas.0906191106. PMC 2868292. PMID 19915151. Lay summary – LiveScience.
- ↑ Jaskelioff, M; Muller, FL; Paik, JH; Thomas, E; Jiang, S; Adams, AC; Sahin, E; Kost-Alimova, M; Protopopov, A; Cadiñanos, J; Horner, JW; Maratos-Flier, E; Depinho, RA (November 2010). "Telomerase reactivation reverses tissue degeneration in aged telomerase deficient mice". Nature. 469 (7328): 102–6. doi:10.1038/nature09603. PMC 3057569. PMID 21113150. Lay summary – news.discovery.com.
- ↑ "Stop, rewind: the scientists slowing the ageing process". BBC News. 2011-01-26.
- ↑ Bernardes de Jesus, B; Vera, E; Schneeberger, K; Tejera, AM; Ayuso, E; Bosch, F; Blasco, MA (August 2012). "Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer". EMBO Molecular Medicine. 4 (8): 691–704. doi:10.1002/emmm.201200245. PMC 3494070. PMID 22585399.
- ↑ Blasco, MA (August 2005). "Telomeres and human disease: ageing, cancer and beyond". Nature Reviews Genetics. 6 (8): 611–22. doi:10.1038/nrg1656. PMID 16136653.
- ↑ Skloot, Rebecca (2010). The Immortal Life of Henrietta Lacks. New York: Broadway Paperbacks. pp. 216, 217. ISBN 978-1-4000-5218-9.
- ↑ Dr. Todd Hennessey, 2016 University at Buffalo
- ↑ Shay, JW; Bacchetti, S (April 1997). "A survey of telomerase activity in human cancer". Eur. J. Cancer. 33 (5): 787–91. doi:10.1016/S0959-8049(97)00062-2. PMID 9282118.
- ↑ Bryan, TM; Englezou, A; Gupta, J; Bacchetti, S; Reddel, RR (September 1995). "Telomere elongation in immortal human cells without detectable telomerase activity". EMBO J. 14 (17): 4240–8. PMC 394507. PMID 7556065.
- ↑ Henson, JD; Neumann, AA; Yeager, TR; Reddel, RR (January 2002). "Alternative lengthening of telomeres in mammalian cells". Oncogene. 21 (4): 598–610. doi:10.1038/sj.onc.1205058. PMID 11850785.
- ↑ Blackburn, EH (February 2005). "Telomeres and telomerase: their mechanisms of action and the effects of altering their functions". FEBS Lett. 579 (4): 859–62. doi:10.1016/j.febslet.2004.11.036. PMID 15680963.
- 1 2 3 Tian, X; Chen, B; Liu, X (March 2013). "Telomere and Telomerase as Targets for Cancer Therapy". Applied Biochemistry and Biotechnology. 160 (5): 906–21. doi:10.1007/s00018-007-6481-8. PMID 17310277.
- ↑ Griffiths, Anthony J. F.; Wesslet, Susan R.; Carroll, Sean B.; Doebley, John (2008). Introduction to Genetic Analysis. W. H. Freeman. ISBN 978-0-7167-6887-6.
- ↑ Williams, SC (January 2013). "No end in sight for telomerase-targeted cancer drugs.". Nat Med. 19 (1): 6. doi:10.1038/nm0113-6. PMID 23295993.
- ↑ Blasco, MA (2001). "Telomeres in Cancer Therapy". J Biomed Biotechnol. 1 (1): 3–4. doi:10.1155/S1110724301000109. PMC 79678. PMID 12488618.
- 1 2 Shay, Jerry W,; Ying, Zou; Hiyama, Eiso; Wright, Woodring E. (2001). "Telomerase and Cancer". Human Molecular Genetics. 10 (7): 677–685. doi:10.1093/hmg/10.7.677. Retrieved June 2015. Check date values in:
|access-date=(help) - ↑ Gul, Ilhami; Dundar, Ozgur; Bodur, Serkan; Tunca, Yusuf; Tutuncu, Levent (2013). "The Status of Telomerase Enzyme Activity in Benign and Malignant Gynecologic Pathologies". Balkan Medical Journal. 30: 287–292. doi:10.5152/balkanmedj.2013.7328. PMC 4115914. PMID 25207121.
- 1 2 Saretzki, Gabriele (2003). "Telomerase inhibition as Cancer Therapy". Cancer Letter. 194 (2): 209–219. doi:10.1016/s0304-3835(02)00708-5. PMID 12757979.
- ↑ Stoyanov, V (2009). "T-loop deletion factor showing speeding aging of Homo telomere diversity and evolution". Rejuvenation Research. 12 (1): 52.
- ↑ Patel, Kunal P.; Robert H., Vonderheide (2004). "Telomerase as a tumor-associated antigen for cancer immunotherapy". Cytotechnology. 45 (1-2): 91–99. doi:10.1007/s10616-004-5132-2. PMC 3449959. PMID 19003246.
- ↑ "Geron's cancer drug shakes off one FDA hold but remains on pause". FierceBiotech. Retrieved 2015-06-28.
- ↑ "J&J bets up to $935M that Geron's drug can shake a checkered past". FierceBiotech. Retrieved 2015-06-28.
- ↑ Johnson, Steven Ross (September 2, 2015). "Experimental blood disorder therapy shows promise in new studies". Modern Healthcare. Retrieved September 2015. Check date values in:
|access-date=(help) - ↑ Epel, ES; Lin, J; Dhabhar, FS; Wolkowitz, OM; Puterman, E; Karan, L; Blackburn, EH (2010). "Dynamics of telomerase activity in response to acute psychological stress". Brain Behav Immun. 24 (4): 531–9. doi:10.1016/j.bbi.2009.11.018. PMC 2856774. PMID 20018236.
- ↑ "Stem cell stages and the origins of colon cancer: a multidisciplinary perspective.". Stem Cell Rev. 1 (3): 243–51. 2005. doi:10.1385/SCR:1:3:243. PMID 17142861.
- ↑ Jacobs TL, et al. (June 2011). "Intensive meditation training, immune cell telomerase activity, and psychological mediators.". nih.gov. 36: 664–81. doi:10.1016/j.psyneuen.2010.09.010. PMID 21035949.
- ↑ Ristow, Michael (2010). "Telomerase deficiency impairs glucose metabolism and insulin secretion" (PDF). Aging. 2 (10): 650–658. PMC 2993795. PMID 20876939.
- ↑ Yamaguchi, H; Calado, RT; Ly, H; Kajigaya, S; Baerlocher, GM; Chanock, SJ; Lansdorp, PM; Young, NS (April 2005). "Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia". N. Engl. J. Med. 352 (14): 1413–24. doi:10.1056/NEJMoa042980. PMID 15814878.
- ↑ Zhang, A; Zheng, C; Hou, M; Lindvall, C; Li, KJ; Erlandsson, F; Björkholm, M; Gruber, A; Blennow, E; Xu, D (April 2003). "Deletion of the Telomerase Reverse Transcriptase Gene and Haploinsufficiency of Telomere Maintenance in Cri du Chat Syndrome". Am. J. Hum. Genet. 72 (4): 940–8. doi:10.1086/374565. PMC 1180356. PMID 12629597.
- ↑ Yamaguchi, H (June 2007). "Mutations of telomerase complex genes linked to bone marrow failures". J Nippon Med Sch. 74 (3): 202–9. doi:10.1272/jnms.74.202. PMID 17625368.
- ↑ Heiss, NS; Knight, SW; Vulliamy, TJ; Klauck, SM; Wiemann, S; Mason, PJ; Poustka, A; Dokal, I (May 1998). "X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions". Nat. Genet. 19 (1): 32–8. doi:10.1038/ng0598-32. PMID 9590285.
- ↑ Vulliamy, TJ; Walne, A; Baskaradas, A; Mason, PJ; Marrone, A; Dokal, I (2005). "Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure". Blood Cells Mol. Dis. 34 (3): 257–63. doi:10.1016/j.bcmd.2004.12.008. PMID 15885610.
- ↑ Vulliamy, T; Marrone, A; Goldman, F; Dearlove, A; Bessler, M; Mason, PJ; Dokal, I (September 2001). "The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita". Nature. 413 (6854): 432–5. doi:10.1038/35096585. PMID 11574891.
- ↑ Marrone, A; Walne, A; Dokal, I (June 2005). "Dyskeratosis congenita: telomerase, telomeres and anticipation". Current Opinion in Genetics & Development. 15 (3): 249–57. doi:10.1016/j.gde.2005.04.004. PMID 15917199.
- ↑ Armanios, M; Chen, JL; Chang, YP; Brodsky, RA; Hawkins, A; Griffin, CA; Eshleman, JR; Cohen, AR; Chakravarti, A; Hamosh, A; Greider, CW (November 2005). "Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita". Proc. Natl. Acad. Sci. U.S.A. 102 (44): 15960–4. doi:10.1073/pnas.0508124102. PMC 1276104. PMID 16247010.
Further reading
- The Immortal Cell, by Michael D. West, Doubleday (2003) ISBN 978-0-385-50928-2
External links
- The Telomerase Database - A Web-based tool for telomerase research.
- Three-dimensional model of telomerase at MUN
- Elizabeth Blackburn's seminars: Telomeres and Telomerase
- Telomerase at the US National Library of Medicine Medical Subject Headings (MeSH)
{kind=link}