Metal-organic framework

Metal-organic frameworks (MOFs) are compounds consisting of metal ions or clusters coordinated to organic ligands to form one-, two-, or three-dimensional structures. They are a subclass of coordination polymers, with the special feature that they are often porous. The organic ligands included are sometimes referred to as "struts", one example being 1,4-benzenedicarboxylic acid (BDC).
More formally, a metal–organic framework is a coordination network with organic ligands containing potential voids. A coordination network is a coordination compound extending, through repeating coordination entities, in one dimension, but with cross-links between two or more individual chains, loops, or spiro-links, or a coordination compound extending through repeating coordination entities in two or three dimensions; and finally a coordination polymer is a coordination compound with repeating coordination entities extending in one, two, or three dimensions.[1]
In some cases, the pores are stable during elimination of the guest molecules (often solvents) and could be used for the storage of gases such as hydrogen and carbon dioxide. Other possible applications of MOFs are in gas purification, in gas separation, in catalysis, as sensors and as supercapacitors.[2]
Structure
MOFs are composed of two major components: a metal ion or cluster of metal ions and an organic molecule called a linker. For this reason, the materials are often referred to as hybrid organic-inorganic materials, however this use of this terminology has recently been explicitly discouraged.[1] The organic units are typically mono-, di-, tri-, or tetravalent ligands.[3] The choice of metal and linker dictates the structure and hence properties of the MOF. For example, the metal's coordination preference influences the size and shape of pores by dictating how many ligands can bind to the metal and in which orientation.
| Dimensionality of Inorganic | |||||
|---|---|---|---|---|---|
| Dimensionality of Organic | 0 | 1 | 2 | 3 | |
| 0 | Molecular Complexes | Hybrid Inorganic Chains | Hybrid Inorganic Layers | 3-D Inorganic Hybrids | |
| 1 | Chain Coordination Polymers | Mixed Inorganic-Organic Layers | Mixed Inorganic-Organic 3-D Framework | ||
| 2 | Layered Coordination Polymer | Mixed Inorganic-Organic 3-D Framework | |||
| 3 | 3-D Coordination Polymers | ||||
To describe and organize the structures of MOFs, a system of nomenclature has been developed. Subunits of a MOF, called secondary building units (SBU), can be described by topologies common to several structures. Each topology, also called a net, is assigned a symbol, consisting of three lower-case letters in bold. MOF-5, for example, has a pcu net. The database of net structures can be found at the Reticular Chemistry Structure Resource.[5] [6]
Common ligands
| Common name | IUPAC name | Chemical formula | Structural formula |
|---|---|---|---|
| Bidentate Carboxylics | |||
| Oxalic acid | ethanedioic acid | HOOC-COOH | 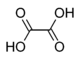 |
| Malonic acid | propanedioic acid | HOOC-(CH2)-COOH |  |
| Succinic acid | butanedioic acid | HOOC-(CH2)2-COOH |  |
| Glutaric acid | pentanedioic acid | HOOC-(CH2)3-COOH | |
| Phthalic acid | benzene-1,2-dicarboxylic acid o-phthalic acid |
C6H4(COOH)2 |  |
| Isophthalic acid | benzene-1,3-dicarboxylic acid m-phthalic acid |
C6H4(COOH)2 |  |
| Terephthalic acid | benzene-1,4-dicarboxylic acid (BDC) p-phthalic acid |
C6H4(COOH)2 |  |
| Biphenyl-4,4'-dicarboxylic acid | Biphenyl-4,4'-dicarboxylic acid (BPDC) | HOOC-(C6H4)2-COOH | |
| Tridentate Carboxylates | |||
| Citric Acid | 2-Hydroxy-1,2,3-propanetricarboxylic acid | (HOOC)CH2C(OH)(COOH)CH2(COOH) | 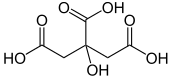 |
| Trimesic acid | benzene-1,3,5-tricarboxylic acid | C9H6O6 |  |
| Azoles | |||
| 1,2,3-Triazole | 1H-1,2,3-triazole | C2H3N3 |  |
| pyrrodiazole | 1H-1,2,4-triazole | C2H3N3 |  |
| Other | |||
| Squaric acid | 3,4-Dihydroxy-3-cyclobutene-1,2-dione | C4H2O4 | 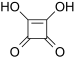 |
Synthesis
The study of MOFs developed from the study of zeolites, except for the use of preformed ligands. MOFs and zeolites are produced almost exclusively by hydrothermal or solvothermal techniques, where crystals are slowly grown from a hot solution. In contrast with zeolites, MOFs are constructed from bridging organic ligands that remain intact throughout the synthesis.[7] Zeolite synthesis often makes use of a "template". Templates are ions that influence the structure of the growing inorganic framework. Typical templating ions are quaternary ammonium cations, which are removed later. In MOFs, the framework is templated by the SBU (secondary building unit) and the organic ligands.[8][9] A templating approach that is useful for MOFs intended for gas storage is the use of metal-binding solvents such as N,N-diethylformamide and water. In these cases, metal sites are exposed when the solvent is evacuated, allowing hydrogen to bind at these sites.[10]
Post-synthetic modification of MOFs opens up possibilities that might not be achieved by conventional synthesis.[11] Of potential relevance to hydrogen storage are MOFs with exposed metal sites. Such sites have been generated by post-synthetic coordination of additional metal ions to sites on the bridging ligands,[10][11] and addition and removal of metal atoms to the metal site.[10][12]
Since ligands in MOFs typically bind reversibly, the slow growth of crystals often allows defects to be redissolved, resulting in a material with millimeter-scale crystals and a near-equilibrium defect density. Solvothermal synthesis is useful for growing crystals suitable to structure determination, because crystals grow over the course of hours to days. However, the use of MOFs as storage materials for consumer products demands an immense scale-up of their synthesis. Scale-up of MOFs has not been widely studied, though several groups have demonstrated that microwaves can be used to nucleate MOF crystals rapidly from solution.[13][14] This technique, termed "microwave-assisted solvothermal synthesis", is widely used in the zeolite literature,[7] and produces micron-scale crystals in a matter of seconds to minutes,[13][14] in yields similar to the slow growth methods.
A solvent-free synthesis of a range of crystalline MOFs has been described. [15] Usually the metal acetate and the organic proligand are mixed and ground up with a ball mill. Cu3(BTC)2 can be quickly synthesised in this way in quantitative yield. In the case of Cu3(BTC)2 the morphology of the solvent free synthesised product was the same as the industrially made Basolite C300. It is thought that localised melting of the components due to the high collision energy in the ball mill may assist the reaction. The formation of acetic acid as a by-product in the reactions in the ball mill may also help in the reaction having a solvent effect[16] in the ball mill.
A recent advancement in the solvent-free preparation of MOF films and composites is their synthesis by chemical vapor deposition. This process, MOF-CVD,[17] was first demonstrated for ZIF-8 and consist of two steps. In a first step, metal oxide precursor layers are deposited. In the second step, these precursor layers are exposed to sublimed ligand molecules, that induce a phase transformation to the MOF crystal lattice. Formation of water during this reaction plays a crucial role in directing the transformation.
Composite materials
Another approach to increasing adsorption in MOFs is to alter the system in such a way that chemisorption becomes possible. This functionality has been introduced by making a composite material, which contains a MOF and a complex of platinum with activated carbon. In an effect known as hydrogen spillover, H2 can bind to the platinum surface through a dissociative mechanism which cleaves the hydrogen molecule into two hydrogen atoms and enables them to travel down the activated carbon onto the surface of the MOF. This innovation produced a threefold increase in the room-temperature storage capacity of a MOF; however, desorption can take upwards of 12 hours, and reversible desorption is sometimes observed for only two cycles.[18][19] The relationship between hydrogen spillover and hydrogen storage properties in MOFs is not well understood but may prove relevant to hydrogen storage.
Hydrogen storage
Since the specific energy of uncompressed hydrogen gas is very low, denser storage methods are of interest for energy storage.[20] MOFs attract attention as materials for adsorptive hydrogen storage because of their high specific surface areas and chemically tunable structures.[18] Hydrogen is taken up by MOFs by adsorption. Compared to an empty gas cylinder, a MOF-filled gas cylinder can store more gas because of adsorption that takes place on the surface of MOFs. (Note that molecular hydrogen adsorbs to the surface, not atomic hydrogen.) Furthermore, MOFs are free of dead-volume, so there is almost no loss of storage capacity as a result of space-blocking by non-accessible volume.[3] Also, MOFs have a fully reversible uptake-and-release behavior: since the storage mechanism is based primarily on physisorption. No large activation barriers are required when liberating the adsorbed hydrogen.[3] The storage capacity of a MOF is limited by the liquid-phase density of hydrogen because the benefits provided by MOFs can be realized only if the hydrogen is in its gaseous state.[3]
In order to realize the benefits provided, such as adsorption, by MOFs hydrogen cannot be stored in them at densities greater than its liquid-phase density.[3] The extent to which a gas can adsorb to a MOF's surface depends on the temperature and pressure of the gas. In general, adsorption increases with decreasing temperature and increasing pressure[3] (until a maximum is reached, typically 20-30 bar, after which the adsorption capacity decreases).,[3][18] However, MOFs to be used for hydrogen storage in automotive fuel cells need to operate efficiently at ambient temperature and pressures between 1 and 100 bar, as these are the values that are deemed safe for automotive applications.[18]
In 2012, MOF-210 was predicted to have hydrogen storage capacity of 2.90 delivery wt% (1-100 bar) at 298 K and 100 bar.[21] Also that MOF-200 will have a Hydrogen storage capacity of 3.24 delivery wt% (1-100 bar) at 298 K and 100 bar.[21] They also proposed new strategies to obtain higher interaction with H2. Such strategy consist on metalating the COF with alkaline metals such as Li.[21] These complexes composed of Li, Na and K bound to benzene ligands (such as 1,3,5-benzenetribenzoate, the ligand used in MOF-177) have been synthesized by Krieck et al.[22] and Goddard showed that the THF is important of their stability. If the metalation with alkaline is performed in the COFs, MOFs are predicted to reach the 2015 DOE target of 5.5 wt % at 298 K: MOF200-Li (6.34 delivery wt%) and MOF200-Na (5.94 6.34 delivery wt%) at 100 bar.[21][23]
Design principles
Practical applications of MOFs for hydrogen storage are met with several challenges. For hydrogen adsorption near room temperature the hydrogen binding energy would need to be increased considerably.[18] Several classes of MOFs have been explored, including carboxylate-based MOFs, heterocyclic azolate-based MOFs, metal-cyanide MOFs, and covalent organic frameworks. Carboxylate-based MOFs have by far received the most attention because
- they are either commercially available or easily synthesized,
- they have high acidity (pKa ˜ 4) allowing for facile in situ deprotonation,
- the metal-carboxylate bond formation is reversible, facilitating the formation of well-ordered crystalline MOFs, and
- the bridging bidentate coordination ability of carboxylate groups favors the high degree of framework connectivity and strong metal-ligand bonds necessary to maintain MOF architecture under the conditions required to evacuate the solvent from the pores.[18]
The most common transition metals employed in carboxylate-based frameworks are Cu2+ and Zn2+. Lighter main group metal ions have also been explored. Be12(OH)12(BTB)4, the first successfully synthesized and structurally characterized MOF consisting of a light main group metal ion, shows high hydrogen storage capacity, but it is too toxic to be employed practically.[24] There is considerable effort being put forth in developing MOFs composed of other light main group metal ions, such as magnesium in Mg4(BDC)3.[18]
The following is a list of several MOFs that are considered to have the best properties for hydrogen storage as of May 2012 (in order of decreasing hydrogen storage capacity).[18] While each MOF described has its advantages, none of these MOFs reach all of the standards set by the U.S. DOE. Therefore, it is not yet known whether materials with high surface areas, small pores, or di- or trivalent metal clusters produce the most favorable MOFs for hydrogen storage.[3]
Zn4O(BTE)(BPDC), where BTE3−=4,4′,4″-[benzene-1,3,5-triyl-tris(ethyne-2,1-diyl)]tribenzoate and BPDC2−=biphenyl-4,4′-dicarboxylate (MOF-210) [25]
Hydrogen storage capacity (at 77 K): 8.6 excess wt% (17.6 total wt%) at 77 K and 80 bar. 44 total g H2/L at 80 bar and 77 K.[25]
Hydrogen storage capacity (at 298 K): 2.90 delivery wt% (1-100 bar) at 298 K and 100 bar.[21]
Zn4O(BBC)2, where BBC3−=4,4′,4″-[benzene-1,3,5-triyl-tris(benzene-4,1-diyl)]tribenzoate (MOF-200) [25]
Hydrogen storage capacity (at 77 K): 7.4 excess wt% (16.3 total wt%) at 77 K and 80 bar. 36 total g H2/L at 80 bar and 77 K.[25]
Hydrogen storage capacity (at 298 K): 3.24 delivery wt% (1-100 bar) at 298 K and 100 bar.[21]
Zn4O(BTB)2, where BTB3−=1,3,5-benzenetribenzoate (MOF-177) [26]
Structure: Tetrahedral [Zn4O]6+ units are linked by large, triangular tricarboxylate ligands. Six diamond-shaped channels (upper) with diameter of 10.8 Å surround a pore containing eclipsed BTB3− moieties (lower).
Hydrogen storage capacity: 7.1 wt% at 77 K and 40 bar; 11.4 wt% at 78 bar and 77 K.
MOF-177 has larger pores, so hydrogen is compressed within holes rather than adsorbed to the surface. This leads to higher total gravimetric uptake but lower volumetric storage density compared to MOF-5.[18]
Zn4O(BDC)3, where BDC2−=1,4-benzenedicarboxylate (MOF-5) [27]
Structure: Square openings are either 13.8 or 9.2 Å depending on the orientation of the aromatic rings.
Hydrogen storage capacity: 7.1 wt% at 77 K and 40 bar ; 10 wt% at 100 bar; volumetric storage density of 66 g/L.
MOF-5 has received much attention from theorists because of the partial charges on the MOF surface, which provide a means of strengthening the binding hydrogen through dipole-induced intermolecular interactions; however, MOF-5 has poor performance at room temperature (9.1 g/L at 100 bar).[18]
Mn3[(Mn4Cl)3(BTT)8]2, where H3BTT=benzene-1,3,5-tris(1H-tetrazole) [28]
Structure: Consists of truncated octahedral cages that share square faces, leading to pores of about 10 Å in diameter. Contains open Mn2+ coordination sites.
Hydrogen storage capacity: 60 g/L at 77 K and 90 bar; 12.1 g/L at 90 bar and 298 K.
This MOF is the first demonstration of open metal coordination sites increasing strength of hydrogen adsorption, which results in improved performance at 298 K. It has relatively strong metal-hydrogen interactions, attributed to a spin state change upon binding or to a classical Coulombic attraction.[18]
Cu3(BTC)2(H2O)3, where H3BTC=1,3,5-benzenetricarboxylic acid [29]
Structure: Consists of octahedral cages that share paddlewheel units to define pores of about 9.8 Å in diameter.
High hydrogen uptake is attributed to overlapping attractive potentials from multiple copper paddle-wheel units: each Cu(II) center can potentially lose a terminal solvent ligand bound in the axial position, providing an open coordination site for hydrogen binding.[18]
Structural impacts on hydrogen storage capacity
To date, hydrogen storage in MOFs at room temperature is a battle between maximizing storage capacity and maintaining reasonable desorption rates, while conserving the integrity of the adsorbent framework (e.g. completely evacuating pores, preserving the MOF structure, etc.) over many cycles. There are two major strategies governing the design of MOFs for hydrogen storage:
- 1) to increase the theoretical storage capacity of the material, and
- 2) to bring the operating conditions closer to ambient temperature and pressure. Rowsell and Yaghi have identified several directions to these ends in some of the early papers.[30][31]
Surface area
The general trend in MOFs used for hydrogen storage is that the greater the surface area, the more hydrogen the MOF can store. High surface area materials tend to exhibit increased micropore volume and inherently low bulk density, allowing for more hydrogen adsorption to occur.[18]
Hydrogen adsorption enthalpy
High hydrogen adsorption enthalpy is also important. Theoretical studies have shown that 22-25 kJ/mol interactions are ideal for hydrogen storage at room temperature, as they are strong enough to adsorb H2, but weak enough to allow for quick desorption.[32] The interaction between hydrogen and uncharged organic linkers is not this strong, and so a considerable amount of work has gone in synthesis of MOFs with exposed metal sites, to which hydrogen adsorbs with an enthalpy of 5-10 kJ/mol. Synthetically, this may be achieved by using ligands whose geometries prevent the metal from being fully coordinated, by removing volatile metal-bound solvent molecules over the course of synthesis, and by post-synthetic impregnation with additional metal cations.[10][28] (C5H5)V(CO)3(H2) and Mo(CO)5(H2) are great examples of increased binding energy due to open metal coordination sites;[33] however, their high metal-hydrogen bond dissociation energies result in a tremendous release of heat upon loading with hydrogen, which is not favorable for fuel cells.[18] MOFs, therefore, should avoid orbital interactions that lead to such strong metal-hydrogen bonds and employ simple charge-induced dipole interactions, as demonstrated in Mn3[(Mn4Cl)3(BTT)8]2.
An association energy of 22-25 kJ/mol is typical of charge-induced dipole interactions, and so there is interest in the use of charged linkers and metals.[18] The metal–hydrogen bond strength is diminished in MOFs, probably due to charge diffusion, so 2+ and 3+ metal ions are being studied to strengthen this interaction even further. A problem with this approach is that MOFs with exposed metal surfaces have lower concentrations of linkers; this makes them difficult to synthesize, as they are prone to framework collapse. This may diminish their useful lifetimes as well.[10][18] Most common strategies to increase this binding energy for MOFs and molecular hydrogen have been reviewed.[23]
Sensitivity to air
MOFs are frequently air/moisture-sensitive. In particular, IRMOF-1 degrades in the presence of small amounts of water at room temperature. Studies on metal analogues have unraveled the ability of metals other than Zn to stand higher water concentrations at high temperatures.[34][35]
To compensate for this, specially constructed storage containers are required, which can be costly. Strong metal-ligand bonds, such as in metal-imidazolate, -triazolate, and -pyrazolate frameworks, are known to decrease a MOF's sensitivity to air, reducing the expense of storage.[18]
Pore size
In a microporous material where physisorption and weak van der Waals forces dominate adsorption, the storage density is greatly dependent on the size of the pores. Calculations of idealized homogeneous materials, such as graphitic carbons and carbon nanotubes, predict that a microporous material with 7 Å-wide pores will exhibit maximum hydrogen uptake at room temperature. At this width, exactly two layers of hydrogen molecules adsorb on opposing surfaces with no space left in between.[18] [36] 10 Å-wide pores are also of ideal size because at this width, exactly three layers of hydrogen can exist with no space in between.[18] (A hydrogen molecule has a bond length of 0.74 Å with a van der Waals radius of 1.17 Å for each atom; therefore, its effective van der Waals length is 3.08 Å.) [37]
Structural defects
Structural defects also play an important role in the performance of MOFs. Room-temperature hydrogen uptake via bridged spillover is mainly governed by structural defects, which can have two effects:
- 1) a partially collapsed framework can block access to pores; thereby reducing hydrogen uptake, and
- 2) lattice defects can create an intricate array of new pores and channels causing increased hydrogen uptake.[38]
Structural defects can also leave metal-containing nodes incompletely coordinated. This enhances the performance of MOFs used for hydrogen storage by increasing the number of accessible metal centers.[39] Finally, structural defects can affect the transport of phonons, which affects the thermal conductivity of the MOF.[40]
Hydrogen adsorption
Adsorption is the process of trapping atoms or molecules that are incident on a surface; therefore the adsorption capacity of a material increases with its surface area. In three dimensions, the maximum surface area will be obtained by a structure which is highly porous, such that atoms and molecules can access internal surfaces. This simple qualitative argument suggests that the highly porous metal-organic frameworks (MOFs) should be excellent candidates for hydrogen storage devices.
Adsorption can be broadly classified as being one of two types: physisorption or chemisorption. Physisorption is characterized by weak van der Waals interactions, and bond enthalpies typically less than 20 kJ/mol. Chemisorption, alternatively, is defined by stronger covalent and ionic bonds, with bond enthalpies between 250 and 500 kJ/mol. In both cases, the adsorbate atoms or molecules (i.e. the particles which adhere to the surface) are attracted to the adsorbent (solid) surface because of the surface energy that results from unoccupied bonding locations at the surface. The degree of orbital overlap then determines if the interactions will be physisorptive or chemisorptive.[41]
Adsorption of molecular hydrogen in MOFs is physisorptive. Since molecular hydrogen only has two electrons, dispersion forces are weak, typically 4-7 kJ/mol, and are only sufficient for adsorption at temperatures below 298 K.[18]
A complete explanation of the H2 sorption mechanism in MOFs was achieved by statistical averaging in the grand canonical ensemble, exploring a wide range of pressures and temperatures.[42][43] Later work revealed a pore filling mechanism as important in both COFs and MOFs.[21]
Determining hydrogen storage capacity
Two hydrogen-uptake measurement methods are used for the characterization of MOFs as hydrogen storage materials: gravimetric and volumetric. To obtain the total amount of hydrogen in the MOF, both the amount of hydrogen absorbed on its surface and the amount of hydrogen residing in its pores should be considered. To calculate the absolute absorbed amount (Nabs), the surface excess amount (Nex) is added to the product of the bulk density of hydrogen (ρbulk) and the pore volume of the MOF (Vpore), as shown in the following equation:[44]
Gravimetric method
The increased mass of the MOF due to the stored hydrogen is directly calculated by a highly sensitive microbalance.[44] Due to buoyancy, the detected mass of adsorbed hydrogen decreases again when a sufficiently high pressure is applied to the system because the density of the surrounding gaseous hydrogen becomes more and more important at higher pressures. Thus, this "weight loss" has to be corrected using the volume of the MOF’s frame and the density of hydrogen.[45]
Volumetric method
The changing of amount of hydrogen stored in the MOF is measured by detecting the varied pressure of hydrogen at constant volume.[44] The volume of adsorbed hydrogen in the MOF is then calculated by subtracting the volume of hydrogen in free space from the total volume of dosed hydrogen.[46]
Other methods of hydrogen storage
There are six possible methods that can be used for the reversible storage of hydrogen with a high volumetric and gravimetric density, which are summarized in the following table, (where ρm is the gravimetric density, ρv is the volumetric density, T is the working temperature, and P is the working pressure):[47]
| Storage method | ρm (mass%) | ρv (kg H2/m3) | T (°C) | P (bar) | Remarks |
|---|---|---|---|---|---|
| High-pressure gas cylinders | 13 | <40 | 25 | 800 | Compressed H2 gas in lightweight composite cylinder |
| Liquid hydrogen in cryogenic tanks | size-dependent | 70.8 | −252 | 1 | Liquid H2; continuous loss of a few percent of H2 per day at 25 °C |
| Adsorbed hydrogen | ~2 | 20 | −80 | 100 | Physisorption of H2 on materials |
| Adsorbed on interstitial sites in a host metal | ~2 | 150 | 25 | 1 | Atomic hydrogen reversibly adsorbs in host metals |
| Complex compounds | <18 | 150 | >100 | 1 | Complex compounds ([AlH4]− or [BH4]−); desorption at elevated temperature, adsorption at high pressures |
| Metal and complexes together with water | <40 | >150 | 25 | 1 | Chemical oxidation of metals with water and liberation of H2 |
Of these, high-pressure gas cylinders and liquid hydrogen in cryogenic tanks are the least practical ways to store hydrogen for the purpose of fuel due to the extremely high pressure required for storing hydrogen gas or the extremely low temperature required for storing hydrogen liquid. The other methods are all being studied and developed extensively.[47]
Catalysis
MOFs have potential as heterogeneous catalysts, although applications have not been commercialized.[48] Their high surface area, tunable porosity, diversity in metal and functional groups make them especially attractive for use as catalysts. Zeolites are extraordinarily useful in catalysis.[49] Zeolites are limited by the fixed tetrahedral coordination of the Si/Al connecting points and the two-coordinated oxide linkers. Fewer than 200 zeolites are known. In contrast with this limited scope, MOFs exhibit more diverse coordination geometries, polytopic linkers, and ancillary ligands (F−, OH−, H2O among others). It is also difficult to obtain zeolites with pore sizes larger than 1 nm, which limits the catalytic applications of zeolites to relatively small organic molecules (typically no larger than xylenes). Furthermore, mild synthetic conditions typically employed for MOF synthesis allow direct incorporation of delicate functionalities into the framework structures. Such a process would not be possible with zeolites or other microporous crystalline oxide-based materials because of the harsh conditions typically used for their synthesis (e.g., calcination at high temperatures to remove organic templates).
Zeolites still cannot be obtained in enantiopure form, which precludes their applications in catalytic asymmetric synthesis, e.g., for the pharmaceutical, agrochemical, and fragrance industries. Enantiopure chiral ligands or their metal complexes have been incorporated into MOFs to lead to efficient asymmetric catalysts. Even some MOF materials may bridge the gap between zeolites and enzymes when they combine isolated polynuclear sites, dynamic host-guest responses, and a hydrophobic cavity environment. MOFs might be useful for making semi-conductors. Theoretical calculations show that MOFs are semiconductors or insulators with band gaps between 1.0 and 5.5 eV which can be altered by changing the degree of conjugation in the ligands indicating its possibility for being photocatalysts.
Despite the many attractions for MOF's, zeolites represent pervasive platforms for industrial catalysis, while MOF's are of no commercial value.
Design
Like other heterogeneous catalysts, MOFs may allow for easier post-reaction separation and recyclability than homogeneous catalysts. In some cases, they also give a highly enhanced catalyst stability. Additionally, they typically offer substrate-size selectivity. Nevertheless, while clearly important for reactions in living systems, selectivity on the basis of substrate size is of limited value in abiotic catalysis, as reasonably pure feedstocks are generally available.
Metal ions or metal clusters

Among the earliest reports of MOF-based catalysis was the cyanosilylation of aldehydes by a 2D MOF (layered square grids) of formula Cd(4,4’-bpy)2(NO3)2.[50] This investigation centered mainly on size- and shape-selective clathration. A second set of examples was based on a two-dimensional, square-grid MOF containing single Pd(II) ions as nodes and 2-hydroxypyrimidinolates as struts.[51] Despite initial coordinative saturation, the palladium centers in this MOFMOF catalyze alcohol oxidation, olefin hydrogenation, and Suzuki C–C coupling. At a minimum, these reactions necessarily entail redox oscillations of the metal nodes between Pd(II) and Pd(0) intermediates accompanying by drastic changes in coordination number, which would certainly lead to destabilization and potential destruction of the original framework if all the Pd centers are catalytically active. The observation of substrate shape- and size-selectivity implies that the catalytic reactions are heterogeneous and are indeed occurring within the MOF. Nevertheless, at least for hydrogenation, it is difficult to rule out the possibility that catalysis is occurring at the surface of MOF-encapsulated palladium clusters/nanoparticles (i.e., partial decomposition sites) or defect sites, rather than at transiently labile, but otherwise intact, single-atom MOF nodes. "Opportunistic" MOF-based catalysis has been described for the cubic compound, MOF-5.[52] This material comprises coordinatively saturated Zn4O nodes and a fully complexed BDC struts (see above for abbreviation); yet it apparently catalyzes the Friedel–Crafts tert-butylation of both toluene and biphenyl. Furthermore, para alkylation is strongly favored over ortho alkylation, a behavior thought to reflect the encapsulation of reactants by the MOF.
Functional struts
The porous-framework material [Cu3(btc)2(H2O)3], also known as HKUST-1,[53] contains large cavities having windows of diameter ~6 Å. The coordinated water molecules are easily removed, leaving open Cu(II) sites. Kaskel and co-workers showed that these Lewis acid sites could catalyze the cyanosilylation of benzaldehyde or acetone. The anhydrous version of HKUST-1 is an acid catalyst.[54] Compared to Brønsted vs. Lewis acid-catalyzed pathways, the product selectivity are distinctive for three reactions: isomerization of a-pinene oxide, cyclization of citronellal, and rearrangement of a-bromoacetals, indicating that indeed [Cu3(btc)2] functions primarily as a Lewis acid catalyst.
MIL-101, a large-cavity MOF having the formula[Cr3F(H2O)2O(BDC)3], is a cyanosilylation catalyst.[55] The coordinated water molecules in MIL-101 are easily removed to expose Cr(III) sites. As one might expect, given the greater Lewis acidity of Cr(III) vs. Cu(II), MIL-101 is much more active than HKUST-1 as a catalyst for the cyanosilylation of aldehydes. Additionally, the Kaskel group observed that the catalytic sites of MIL-101, in contrast to those of HKUST-1, are immune to unwanted reduction by benzaldehyde. The Lewis-acid-catalyzed cyanosilylation of aromatic aldehydes has also been carried out by Long and co-workers using a MOF of the formula Mn3[(Mn4Cl)3BTT8(CH3OH)10].[56] This material contains a three-dimensional pore structure, with the pore diameter equaling 10 Å. In principle, either of the two types of Mn(II) sites could function as a catalyst. Noteworthy features of this catalyst are high conversion yields (for small substrates) and good substrate-size-selectivity, consistent with channellocalized catalysis.
Encapsulated catalysts
The MOF encapsulation approach invites comparison to earlier studies of oxidative catalysis by zeolite-encapsulated Fe(porphyrin)[57] as well as Mn(porphyrin)[58] systems. The zeolite studies generally employed iodosylbenzene (PhIO), rather than TPHP as oxidant. The difference is likely mechanistically significant, thus complicating comparisons. Briefly, PhIO is a single oxygen atom donor, while TBHP is capable of more complex behavior. In addition, for the MOF-based system, it is conceivable that oxidation proceeds via both oxygen transfer from a manganese oxo intermediate as well as a manganese-initiated radical chain reaction pathway. Regardless of mechanism, the approach is a promising one for isolating and thereby stabilizing the porphyrins against both oxo-bridged dimer formation and oxidative degradation.[59]
Metal-free organic cavity modifiers
Most examples of MOF-based catalysis make use of metal ions or atoms as active sites. Among the few exceptions are two nickel- and two copper-containing MOFs synthesized by Rosseinsky and co-workers.[60] These compounds employ amino acids(L- or D-aspartate) together with dipyridyls as struts. The coordination chemistry is such that the amine group of the aspartate cannot be protonated by added HCl, but one of the aspartate carboxylates can. Thus, the framework-incorporated amino acid can exist in a form that is not accessible for the free amino acid. While the nickel-based compounds are marginally porous, on account of tiny channel dimensions, the copper versions are clearly porous. The Rosseinsky group showed that the carboxylic acids behave as Brønsted acidic catalysts, facilitating (in the copper cases) the ring-opening methanolysis of a small, cavityaccessible epoxide at up to 65% yield. These researchers point out that superior homogeneous catalysts exist, but emphasize that the catalyst formed here is unique to the MOF environment, thus representing an interesting proof of concept. Kitagawa and co-workers have reported the synthesis of a catalytic MOF having the formula [Cd(4-BTAPA)2(NO3)2].[61] The MOF is three-dimensional, consisting of an identical catenated pair of networks, yet still featuring pores of molecular dimensions. The nodes consist of single cadmium ions, octahedrally ligated by pyridyl nitrogens. From a catalysis standpoint, however, the most interesting feature of this material is the presence of guest-accessible amide functionalities. The researchers showed that the amides are capable of base-catalyzing the Knoevenagel condensation of benzaldehyde with malononitrile. Reactions with larger nitriles, however, are only marginally accelerated, implying that catalysis takes place chiefly within the material’s channels rather than on its exterior. A noteworthy finding is the lack of catalysis by the free strut in homogeneous solution, evidently due to intermolecular H-bonding between bptda molecules. Thus, the MOF architecture elicits catalytic activity not otherwise encountered. In an interesting alternative approach, Férey and coworkers were able to modify the interior of MIL-101 via Cr(III) coordination of one of the two available nitrogen atoms of each of several ethylenediamine molecules.[62] The free non-coordinated ends of the ethylenediamines were then used as Brønsted basic catalysts, again for Knoevenagel condensation of benzaldehyde with nitriles. A third approach has been described by Kim and coworkers.[63] Using a pyridine-functionalized derivative of tartaric acid and a Zn(II) source they were able to synthesize a 2D MOF termed POST-1. POST-1 possesses 1D channels whose cross sections are defined by six trinuclear zinc clusters and six struts. While three of the six pyridines are coordinated by zinc ions, the remaining three are protonated and directed toward the channel interior. When neutralized, the noncoordinated pyridyl groups are found to catalyze transesterification reactions, presumably by facilitating deprotonation of the reactant alcohol. The absence of significant catalysis when large alcohols are employed strongly suggests that the catalysis occurs within the channels of the MOF.
Achiral catalysis
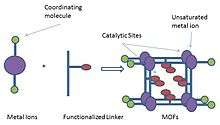
Metals as catalytic sites
The metals in the MOF structure often act as Lewis acids. The metals in MOFs often coordinate to labile solvent molecules or counter ions which can be removed after activation of the framework. The Lewis acidic nature of such unsaturated metal centers can activate the coordinated organic substrates for subsequent organic transformations. The use of unsaturated metal centers was demonstrated in the cyanosilylation of aldehydes and imines by Fujita and coworkers in 2004.[64] They reported MOF of composition {[Cd(4,4′-bpy)2(H2O)2] • (NO3)2 • 4H2O} which was obtained by treating linear bridging ligand 4,4′-bipyridine (bpy) with Cd(NO3)2 . The Cd(II) centers in this MOF possesses a distorted octahedral geometry having four pyridines in the equatorial positions, and two water molecules in the axial positions to form a two-dimensional infinite network. On activation, two water molecules were removed leaving the metal centers unsaturated and Lewis acidic. The Lewis acidic character of metal center was tested on cyanosilylation reactions of imine where the imine gets attached to the Lewis-acidic metal centre resulting in higher electrophilicity of imines. For the cyanosilylation of imines, most of the reactions were complete within 1 h affording aminonitriles in quantitative yield. Kaskel and coworkers[65] carried out similar cyanosilylation reactions with coordinatively unsaturated metals in three-dimensional (3D) MOFs as heterogeneous catalysts. The 3D framework [Cu3(btc)2(H2O)3] (btc: Benzene-1,3,5- tricarboxylate) (HKUST-1) used in this study was first reported by Williams et al.[66] The open framework of [Cu3(btc)2(H2O)3] is built from dimeric cupric tetracarboxylate units (paddle-wheels) with aqua molecules coordinating to the axial positions and btc bridging ligands. The resulting framework after removal of two water molecules from axial positions possesses porous channel. This activated MOF catalyzes the trimethylcyanosilylation of benzaldehydes with a very low conversion (<5% in 24 h) at 293 K. As the reaction temperature was raised to 313 K, a good conversion of 57% with a selectivity of 89% was obtained after 72 h. In comparison, less than 10% conversion was observed for the background reaction (without MOF) under the same conditions. But this strategy suffers from some problems like 1) the decomposition of the framework with increase of the reaction temperature due to the reduction of Cu(II) to Cu(I) by aldehydes; 2) strong solvent inhibition effect; electron donating solvents such as THF competed with aldehydes for coordination to the Cu(II) sites, and no cyanosilylation product was observed in these solvents; 3) the framework instability in some organic solvents. Several other groups have also reported the use of metal centres in MOFs as catalysts[67][68] Again, electron-deficient nature of some metals and metal clusters makes the resulting MOFs efficient oxidation catalysts. Mori and coworkers[69] reported MOFs with Cu2 paddle wheel units as heterogeneous catalysts for the oxidation of alcohols. The catalytic activity of the resulting MOF was examined by carrying out alcohol oxidation with H2O2 as the oxidant. It also catalyzed the oxidation of primary alcohol, secondary alcohol and benzyl alcohols with high selectivity. Hill et al.[70] have demonstrated the sulfoxidation of thioethers using an MOF based on vanadium-oxo cluster V6O13 building units.
Functional linkers as catalytic sites
Functional linkers can be also utilized as catalytic sites. A 3D MOF {[Cd(4- BTAPA)2(NO3)2] • 6H2O • 2DMF} (4-BTAPA=1,3,5-benzene tricarboxylic acid tris [N-(4-pyridyl)amide], DMF=N,N-dimethylformamide) constructed by tridentate amide linkers and cadmium salt catalyzes the Knoevenagel condensation reaction.[71] The pyridine groups on the ligand 4-BTAPA act as ligands binding to the octahedral cadmium centers, while the amide groups can provide the functionality for interaction with the incoming substrates. Specifically, the – NH moiety of the amide group can act as electron acceptor whereas the C=O group can act as electron donor to activate organic substrates for subsequent reactions. Ferey et al.[72] reported a robust and highly porous MOF [Cr3(µ3-O)F(H2O)2(BDC)3] (BDC: Benzene-1,4- dicarboxylate) where instead of directly using the unsaturated Cr(III) centers as catalytic sites, the authors grafted ethylenediamine (ED) onto the Cr(III) sites. The uncoordinated ends of ED can act as base catalytic sites, ED-grafted MOF was investigated for Knoevenagel condensation reactions. A significant increase in conversion was observed for ED-grafted MOF compared to untreated framework (98% vs 36%).
Entrapment of catalytically active noble metal nanoparticles
The entrapment of catalytically active noble metals can be accomplished by grafting on functional groups to the unsaturated metal site on MOFs. Ethylenediamine (ED) has been shown to be grafted on the Cr metal sites and can be further modified to encapsulate noble metals such as Pd.[73] The entraped Pd has similar catalytic activity as Pd/C in the Heck reaction. Ruthenium nanoparticles have catalytic activity in a number of reactions when entrapped in the MOF-5 framework.[74] This Ru-encapsulated MOF catalyzes oxidation of benzyl alcohol to benzyaldehyde, although degradation of the MOF occurs. The same catalyst was used in the hydrogenation of benzene to cyclohexane.
Reaction hosts with size selectivity
MOFs might prove useful for both photochemical and polymerization reactions due to the tuneability of the size and shape of their pores. A 3D MOF {[Co(bpdc)3(bpy)] • 4DMF • H2O} (bpdc: biphenyldicarboxylate, bpy: 4,4′-bipyridine) was synthesized by Li and coworkers.[75] Using this MOF photochemistry of o-methyl dibenzyl ketone (o-MeDBK) was extensively studied. This molecule was found to have a variety of photochemical reaction properties including the production of cyclopentanol. MOFs have been used to study polymerization in the confined space of MOF channels. Polymerization reactions in confined space might have different properties than polymerization in open space. Styrene, divinylbenzene, substituted acetylenes, methyl methacrylate, and vinyl acetate have all been studied by Kitagawa and coworkers as possible activated monomers for radical polymerization.[76][77] Due to the different linker size the MOF channel size could be tunable on the order of roughly 25 and 100 Å2. The channels were shown to stabilize propagating radicals and suppress termination reactions when used as radical polymerization sites.
Asymmetric catalysis
Several strategies exist for constructing homochiral MOFs. Crystallization of homochiral MOFs via self-resolution from achiral linker ligands is one of the way to accomplish such a goal. However, the resulting bulk samples contain both enantiomorphs and are racemic. Aoyama and coworkers[78] successfully obtained homochiral MOFs in the bulk from achiral ligands by carefully controlling nucleation in the crystal growth process. Zheng and coworkers[79] reported the synthesis of homochiral MOFs from achiral ligands by chemically manipulating the statistical fluctuation of the formation of enantiomeric pairs of crystals. Growing MOF crystals under chiral influences is another approach to obtain homochiral MOFs using achiral linker ligands. Rosseinsky and coworkers[80][81] have introduced a chiral coligand to direct the formation of homochiral MOFs by controlling the handedness of the helices during the crystal growth. Morris and coworkers[82] utilized ionic liquid with chiral cations as reaction media for synthesizing MOFs, and obtained homochiral MOFs. The most straightforward and rational strategy for synthesizing homochiral MOFs is, however, to use the readily available chiral linker ligands for their construction.
Homochiral MOFs with interesting functionalities and reagent-accessible channels
Homochiral MOFs have been made by Lin and coworkers using 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), 1,1′-bi-2,2′-naphthol (BINOL) as a chiral ligands.[83] These ligands can coordinate with catalytically active metal sites to enhance the enantioselectivity. A variety of linking groups such as pyridine, phosphonic acid, and carboxylic acid can be selectively introduced to the 3,3′, 4,4′, and the 6,6′ positions of the 1,1'-binaphthyl moiety. Moreover, by changing the length of the linker ligands the porosity and framework structure of the MOF can be selectivily tuned.
Postmodification of homochiral MOFs
Lin and coworkers have shown that the postmodification of MOFs can be achieved to produce enantioselective homochiral MOFs for use as catalysts.[84] The resulting 3D homochiral MOF {[Cd3(L)3Cl6]• 4DMF • 6MeOH • 3H2O} (L=(R)-6,6'-dichloro-2,2'-dihydroxyl-1,1'-binaphthyl-bipyridine) synthesized by Lin was shown to have a similar catalytic efficiency for the diethylzinc addition reaction as compared to the homogeneous analogue when was pretreated by Ti(OiPr)4 to generate the grafted Ti- BINOLate species. The catalytic activity of MOFs can vary depending on the framework structure. Lin and others found that MOFs synthesized from the same materials could have drastically different catalytic activities depending on the framework structure present.[85]
Homochiral MOFs with precatalysts as building blocks
Another approach to construct catalytically active homochiral MOFs is to incorporate chiral metal complexes which are either active catalysts or precatalysts directly into the framework structures. For example, Hupp and coworkers[86] have combined a chiral ligand and bpdc (bpdc: biphenyldicarboxylate) with Zn(NO3)2 and obtained twofold interpenetrating 3D networks. The orientation of chiral ligand in the frameworks makes all Mn(III) sites accessible through the channels. The resulting open frameworks showed catalytic activity towards asymmetric olefin epoxidation reactions. No significant decrease of catalyst activity was observed during the reaction and the catalyst could be recycled and reused several times. Lin and coworkers[87] have reported zirconium phosphonate-derived Ru-BINAP systems. Zirconium phosphonate-based chiral porous hybrid materials containing the Ru(BINAP)(diamine)Cl2 precatalysts showed excellent enantioselectivity (up to 99.2% ee) in the asymmetric hydrogenation of aromatic ketones.
Biomimetic design and photocatalysis
Some MOF materials may resemble enzymes when they combine isolated polynuclear sites, dynamic host-guest responses, and hydrophobic cavity environment which are characteristics of an enzyme.[88] Some well-known examples of cooperative catalysis involving two metal ions in biological systems include: the diiron sites in methane monooxygenase, dicopper in cytochrome c oxidase, and tricopper oxidases which have analogy with polynuclear clusters found in the 0D coordination polymers, such as binuclear Cu2 paddlewheel units found in MOP-1[89][90] and [Cu3(btc)2] (btc=benzene-1,3,5-tricarboxylate) in HKUST-1 or trinuclear units such as {Fe3O(CO2)6} in MIL-88,[91] and IRMOP-51.[92] Thus, 0D MOFs have accessible biomimetic catalytic centers. In enzymatic systems, protein units show "molecular recognition", high affinity for specific substrates. It seems that molecular recognition effects are limited in zeolites by the rigid zeolite structure.[93] In contrast, dynamic features and guest-shape response make MOFs more similar to enzymes. Indeed, many hybrid frameworks contain organic parts that can rotate as a result of stimuli, such as light and heat.[94] The porous channels in MOF structures can be used as photocatalysis sites. In photocatalysis, the use of mononuclear complexes is usually limited either because they only undergo single- electron process or from the need for high-energy irradiation. In this case, binuclear systems have a number of attractive features for the development of photocatalysts.[95] For 0D MOF structures, polycationic nodes can act as semiconductor quantum dots which can be activated upon photostimuli with the linkers serving as photon antennae.[96] Theoretical calculations show that MOFs are semiconductors or insulators with band gaps between 1.0 and 5.5 eV which can be altered by changing the degree of conjugation in the ligands.[97] Experimental results show that the band gap of IRMOF-type samples can be tuned by varying the functionality of the linker.[98]
Additional potential applications
Many potential uses of MOFs exist beyond hydrogen storage, such as gas purification, gas separation, gas storage (other than hydrogen), and heterogeneous catalysis. MOFs are promising for gas purification because of strong chemisorption that takes place between electron-rich, odor-generating molecules (such as amines, phosphines, oxygenates, alcohols, water, or sulfur-containing molecules) and the framework, allowing the desired gas to pass through the MOF. Gas separation can be performed with MOFs because they can allow certain molecules to pass through their pores based on size and kinetic diameter. This is particularly important for separating out carbon dioxide. Regarding gas storage, MOFs can store molecules such as carbon dioxide, carbon monoxide, methane, and oxygen due to their high adsorption enthalpies (similar to hydrogen). In principle, these same MOFs could also be used for catalysis because of their shape and size selectivity and their accessible bulk volume. Also, because of their very porous architecture, mass transport in the pores is not hindered.[3]
Drug delivery systems
As MOFs are now available with a wide range of chemical compositions, many may be toxicologically acceptable for use in drug delivery.[99]
Methane storage
The total uptake and delivery amount in COF-102 and COF-103 outperform other 2D and 3D-COFs at high pressure, even the benchmark MOF-177.[100] In October 2011, new COFs with better delivery amount have been shown to be stable and overcome the DOE target in delivery basis. COF-103-Eth-trans and COF-102-Ant, are found to exceed the DOE target of 180 v(STP)/v at 35 bar for methane storage. Thus at 35 bar (in v(STP)/v delivery units) the best performers is COF-103-Eth-trans which stores 5.6 times as much as bulk CH4 at the same pressure (bulk CH4 reaches 34). All the designed COFs have superior performance to previously reported COFs and MOFs, such as COF-102 (137), MOF-177 (112), and MOF-200 (81 v(STP)/v delivery units).[101] The new materials were designed for best performance at 35 bar. They reported that using thin vinyl bridging groups aid performance by minimizing the interaction methane-COF at low pressure. This can be extended to MOFs. This is a new feature that can be used to enhance loading in addition to the common practice of adding extra fused benzene rings.[101]
Semiconductors
In 2014 researchers proved that they can create electrically conductive thin films of MOFs (Cu3(BTC)2 (also known as HKUST-1; BTC, benzene-1,3,5-tricarboxylic acid) infiltrated with the molecule 7,7,8,8-tetracyanoquinododimethane) that could be used in applications including photovoltaics, sensors and electronic materials and a path towards creating semiconductors. The simulated matrices of 600 atoms.[102] The team demonstrated tunable, air-stable electrical conductivity over six orders of magnitude, with values as high as 7 siemens per meter.[103]
In 2014 Ni
3(2,3,6,7,10,11-hexaiminotriphenylene)2 was shown to be a metal-organic graphene analogue that has a natural 2 nm band gap and is able to self-assemble. It represents a family of similar compounds. Because of the symmetry and geometry in HITP, the overall organometallic complex has an almost fractal nature that allows it to perfectly self-organize. By contrast, graphene must be doped to give it the properties of a semiconductor. Ni3(HITP)2 pellets had a conductivity of 2 S/cm, a record for a metal-organic compound. In 2D sheets, conductance measured 40 S/cm, also a record and among the best for any polymer.[104][105]
Bio-mimetic mineralization
In 2015 researchers at CSIRO proved that they could encapsulate biomolecules during the MOF crystallization process. Biomolecules including proteins and nucleic acids could be encapsulated, and enzymes encapsulated in this way were stable and active even after being treated in harsh reagents and conditions. ZIF-8, MIL-88A, HKUST-1, and several luminescent MOFs containing lanthanide metals were used for the biomimetic mineralization process.[106][107]
Carbon capture
Because of their small, tunable pore sizes and high void fractions, MOFs are a promising potential material for use as an adsorbent to capture CO2. MOFs could provide a more efficient alternative to traditional amine solvent-based methods in CO2 capture from coal-fired power plants.[108]
MOFs could be employed in each of the main three carbon capture configurations for coal-fired power plants: pre-combustion, post-combusiton, and oxy-combustion.[109] However, since the post-combustion configuration is the only one that can be retrofitted to existing plants, it garners the most interest and research. In post-combustion carbon capture, the flue gas from the power plant would be fed through a MOF in a packed-bed reactor setup. Flue gas is generally 40 to 60 °C with a partial pressure of CO2 at 0.13 - 0.16 bar. The main component CO2 must be separated from is nitrogen, but there are small amounts of other gasses as well. A typical flue gas composition is: 73-77% N2, 15-16% CO2, 5-7% H2O, 3-4% O2, 800 ppm SO2, 10 ppm SO3, 500 ppm NOX, 100 ppm HCl, 20 ppm CO, 10 ppm hydrocarbons, 1ppb Hg.[109]
CO2 can bind to the MOF surface through either physisorption or chemisorption, where physisorption occurs through van der Waals interactions and chemisorption occurs through covalent bonds being formed between the CO2 and MOF surface.[110] Once the MOF is saturated with CO2, the MOF would then be regenerated (CO2 is removed from the MOF to be transported elsewhere for sequestration or enhanced oil recovery) through either a temperature swing or a pressure swing. In a temperature swing, the MOF would be heated up until CO2 desorbs; temperature swing regeneration is usually used in post-combustion configurations where the heat is supplied from heat exchangers with the power plant. To achieve working capacities comparable to the amine solvent process, the MOF must be heated up to around 200 C. In a pressure swing, the pressure would be decreased until CO2 desorbs; this would usually be used in a pre-combustion carbon capture configuration.[111]
MOF-177 exhibits a CO2 capacity of 33.5 mmol CO2 / g MOF at ambient temperature and 35 bar. For comparison, zeolite 13X has a CO2 capacity of 7.4 mmol/g at 32 bar and ambient temperature, and MAXSORB has a capacity of 25 mmol/g at 35 bar and ambient temperature. MOF-177 has pore sizes of 11 and 17 Angstroms and consists of zinc complexes connected by benzene rings, with the repeating structure being Zn4O(1,3,5-benzenetribenzoate)2.[112]
In addition to their tunable selectivities for different molecules, another property of MOFs that makes them a good candidate for carbon capture is their low heat capacities. Monoethanolamine (MEA) solutions, the leading method for capturing CO2 from flue gas, have a heat capacity between 3-4 J/g K since they are mostly water. This high heat capacity contributes to the energy penalty in the solvent regeneration step, i.e., when the absorbed CO2 is removed from the MEA solution. MOF-177, on the other hand, has a heat capacity of 0.5 J/g K at ambient temperature.[109]
In a collaborative project sponsored by the U.S. DOE, MOFs were shown to separate 90% of the CO2 from the flue gas stream using a vacuum pressure swing process. The MOF Mg(dobdc) has a 21.7 wt% CO2 loading capacity. Estimations showed that, if a similar system would be applied to a large scale power plant, the cost of energy would increase by 65%, while a U.S. NETL baseline amine-based system would cause an increase of 81% (the U.S. DOE goal is 35%). The cost of capturing CO2 would be $57 / ton CO2 captured, while for the amine system the cost is estimated to be $72 / ton CO2 captured. The project estimated that the total capital required to implement such project in a 580 MW power plant would be $354 million.[113]
See also
- BET theory
- Conjugated microporous polymer
- Coordination chemistry
- Coordination polymers
- Cryogenics
- Crystal nets (periodic graphs)
- Hydrogen
- Hydrogen economy
- Liquid hydrogen
- Macromolecular assembly
- Omar M. Yaghi
- Organometallic chemistry
- United States Department of Energy
- X-ray Crystallography
- Zeolitic imidazolate frameworks
- Solid sorbents for carbon capture
References
- 1 2 Batten, SR; Champness, NR; Chen, X-M; Garcia-Martinez, J; Kitagawa, S; Öhrström, L; O'Keeffe, M; Suh, MP; Reedijk, J (2013). "Terminology of metal–organic frameworks and coordination polymers (IUPAC Recommendations 2013)" (PDF). Pure and Applied Chemistry. 85 (8): 1715. doi:10.1351/PAC-REC-12-11-20.
- ↑ Cejka, J, ed. (2011). Metal-Organic Frameworks Applications from Catalysis to Gas Storage. Wiley-VCH. ISBN 978-3-527-32870-3.
- 1 2 3 4 5 6 7 8 9 Czaja, Alexander U.; Trukhan, Natalia; Müller, Ulrich (2009). "Industrial applications of metal-organic frameworks". Chemical Society Reviews. 38 (5): 1284–1293. doi:10.1039/b804680h. PMID 19384438.
- ↑ Cheetham, AK; Rao, CNR; Feller, RK (2006). "Structural diversity and chemical trends in hybrid inorganic–organic framework materials". Chemical Communications (46): 4780. doi:10.1039/b610264f.
- ↑ Tranchemontagne, David J; Mendoza-Cortes, Jose L; O’Keeffe, Michael; Yaghi, Omar M (2009). "Secondary building units, nets and bonding in the chemistry of metal–organic frameworks". Chemical Society Reviews. 38 (5): 1257–1283. doi:10.1039/B817735J.
- ↑ Tranchemontagne, David J; Mendoza-Cortes, Jose L; O’Keeffe, Michael; Yaghi, Omar M (2009). "Secondary building units, nets and bonding in the chemistry of metal–organic frameworks".
- 1 2 Cheetham, AK; Férey, G; Loiseau, T (1999). "Open-framework inorganic materials". Angewandte Chemie International Edition. 38 (22): 3268–3292. doi:10.1002/(SICI)1521-3773(19991115)38:22<3268::AID-ANIE3268>3.0.CO;2-U. PMID 10602176.
- ↑ Bucar, D-K; Papaefstathiou, GS; Hamilton, TD; Chu, QL; Georgiev, IG; MacGillivray, LR (2007). "Template-controlled reactivity in the organic solid state by principles of coordination-driven self-assembly". European Journal of Inorganic Chemistry. 2007 (29): 4559–4568. doi:10.1002/ejic.200700442.
- ↑ Parnham, ER; Morris, RE (2007). "Ionothermal Synthesis of Zeolites, Metal-Organic Frameworks, and Inorganic-Organic Hybrids". Accounts of Chemical Research. 40 (10): 1005–1013. doi:10.1021/ar700025k. PMID 17580979.
- 1 2 3 4 5 Dinca, Mircea; Long, Jeffrey R. (2008). "Hydrogen storage in microporous metal-organic frameworks with exposed metal sites". Angewandte Chemie International Edition. 47 (36): 6766–6779. doi:10.1002/anie.200801163. PMID 18688902.
- 1 2 Wang, Z; Cohen, SM (2009). "Postsynthetic modification of metal–organic frameworks". Chemical Society Reviews. 38 (5): 1315–1329. doi:10.1039/b802258p. PMID 19384440.
- ↑ Das, S; Kim, H; Kim, K (2009). "Metathesis in Single Crystal: Complete and Reversible Exchange of Metal Ions Constituting the Frameworks of Metal-Organic Frameworks". Journal of the American Chemical Society. 131 (11): 3814–3815. doi:10.1021/ja808995d. PMID 19256486.
- 1 2 Ni, Z; Masel, RI (2006). "Rapid production of metal-organic frameworks via microwave-assisted solvothermal synthesis". Journal of the American Chemical Society. 128 (38): 12394–12395. doi:10.1021/ja0635231. PMID 16984171.
- 1 2 Choi, J-S; Son, W-J; Kim, J; Ahn, W-S (2008). "Metal–organic framework MOF-5 prepared by microwave heating: Factors to be considered". Microporous and Mesoporous Materials. 116 (1–3): 727–731. doi:10.1016/j.micromeso.2008.04.033.
- ↑ Pichon, A; James, SL (2008). "An array-based study of reactivity under solvent-free mechanochemical conditions—insights and trends". CrystEngComm. 10 (12): 1839–1847. doi:10.1039/B810857A.
- ↑ Braga, D; Giaffreda, SL; Grepioni, F; Chierotti, MR; Gobetto, R; Palladino, G; Polito, M (2007). "Solvent effect in a "solvent free" reaction". CrystEngComm. 9 (10): 879–881. doi:10.1039/B711983F.
- ↑ Stassen, I; Styles, M; Grenci, G; Van Gorp, H; Vanderlinden, W; De Feyter, S; Falcaro, P; De Vos, D; Vereecken, P; Ameloot, R (2015). "Chemical vapour deposition of zeolitic imidazolate framework thin films". Nature Materials. doi:10.1038/nmat4509.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 Murray, Leslie J.; Dinca, Mircea; Long, Jeffrey R. (2009). "Hydrogen storage in metal-organic frameworks". Chemical Society Reviews. 38 (5): 1294–1314. doi:10.1039/b802256a. PMID 19384439.
- ↑ Li, Y; Yang, RT (2007). "Hydrogen Storage on Platinum Nanoparticles Doped on Superactivated Carbon". Journal of Physical Chemistry C. 111 (29): 11086–11094. doi:10.1021/jp072867q.
- ↑ Committee on Alternatives and Strategies for Future Hydrogen Production and Use, National Research Council, National Academy of Engineering (2004). The Hydrogen Economy: Opportunities, Costs, Barriers, and R&D Needs. Washington, D.C.: National Academies. pp. 11–24, 37–44. ISBN 0-309-09163-2.
- 1 2 3 4 5 6 7 Mendoza-Cortés, José L.; Han, Sang Soo; Goddard Wa, William A. (2012). "High H2Uptake in Li-, Na-, K-Metalated Covalent Organic Frameworks and Metal Organic Frameworks at 298 K". The Journal of Physical Chemistry A. 116 (6): 1621–31. doi:10.1021/jp206981d. PMID 22188543.
- ↑ Krieck, Sven; GöRls, Helmar; Westerhausen, Matthias (2010). "Alkali Metal-Stabilized 1,3,5-Triphenylbenzene Monoanions: Synthesis and Characterization of the Lithium, Sodium, and Potassium Complexes†". Organometallics. 29 (24): 6790. doi:10.1021/om1009632.
- 1 2 Han, Sang Soo; Mendoza-Cortés, José L.; Goddard Wa, William A. (2009). "Recent advances on simulation and theory of hydrogen storage in metal–organic frameworks and covalent organic frameworks". Chemical Society Reviews. 38 (5): 1460–76. doi:10.1039/B802430H. PMID 19384448.
- ↑ Sumida, Kenji; Hill, Matthew R.; Horike, Satoshi; Dailly, Anne; Long, Jeffrey R. (2009). "Synthesis and hydrogen storage properties of Be12(OH)12(1,3,5-benzenetribenzoate)4". Journal of the American Chemical Society. 131 (42): 15120–15121. doi:10.1021/ja9072707. PMID 19799422.
- 1 2 3 4 Furukawa, H.; Ko, N.; Go, Y. B.; Aratani, N.; Choi, S. B.; Choi, E.; Yazaydin, A. O.; Snurr, R. Q.; O'Keeffe, M.; Kim, J.; Yaghi, O. M. (2010). "Ultrahigh Porosity in Metal-Organic Frameworks". Science. 329 (5990): 424–8. Bibcode:2010Sci...329..424F. doi:10.1126/science.1192160. PMID 20595583.
- ↑ Rowsell, Jesse L. C.; Millward, Andrew R.; Park, Kyo Sung; Yaghi, Omar M. (2004). "Hydrogen sorption in functionalized metal-organic frameworks". Journal of the American Chemical Society. 126 (18): 5666–5667. doi:10.1021/ja049408c. PMID 15125649.
- ↑ Rosi, Nathaniel L.; Eckert, Juergen; Eddaoudi, Mohamed; Vodak, David T.; Kim, Jaheon; O'Keefe, Michael; Yaghi, Omar M. (2003). "Hydrogen storage in microporous metal-organic frameworks". Science. 300 (5622): 1127–1129. Bibcode:2003Sci...300.1127R. doi:10.1126/science.1083440. PMID 12750515.
- 1 2 Dinca, Mircea; Dailly, Anne; Liu, Yun; Brown, Craig M.; Neumann, Dan A.; Long, Jeffrey R. (2006). "Hydrogen storage in a microporous metal-organic framework with exposed Mn2+ coordination sites". Journal of the American Chemical Society. 128 (51): 16876–16883. doi:10.1021/ja0656853. PMID 17177438.
- ↑ Lee, JeongYong; Li, Jing; Jagiello, Jacek (2005). "Gas sorption properties of microporous metal organic frameworks". Journal of Solid State Chemistry. 178 (8): 2527–2532. Bibcode:2005JSSCh.178.2527L. doi:10.1016/j.jssc.2005.07.002.
- ↑ Rowsell, Jesse L. C.; Yaghi, Omar M. (2005). "Strategies for hydrogen storage in metal-organic frameworks". Angewandte Chemie International Edition. 44 (30): 4670–4679. doi:10.1002/anie.200462786. PMID 16028207.
- ↑ Rowsell, Jesse L. C.; Yaghi, Omar M. (2006). "Effects of functionalization, catenation, and variation of the metal oxide and organic linking units on the low-pressure hydrogen adsorption properties of metal-organic frameworks". Journal of the American Chemical Society. 128 (4): 1304–1315. doi:10.1021/ja056639q. PMID 16433549.
- ↑ Garrone, E; Bonelli, B; Arean, C. O. (2008). "Enthalpy-entropy correlation for hydrogen adsorption on zeolites". Chemical Physics Letters. 456 (1–3): 68–70. Bibcode:2008CPL...456...68G. doi:10.1016/j.cplett.2008.03.014.
- ↑ Kubas, G. J. (2001). Metal Dihydrogen and s-Bond Complexes: Structure, Theory, and Reactivity. New York: Kluwer Academic.
- ↑ Lopez, Nuria; Calero, Sofía; López, Núria (2012). "Early stages in the degradation of Metal-Organic Frameworks in liquid water from first-principles Molecular Dynamics". Physical Chemistry Chemical Physics. 14 (20): 7240–7245. Bibcode:2012PCCP...14.7240B. doi:10.1039/C2CP40339K. PMID 22513503.
- ↑ Lopez, Nuria; Castillo, Juan Manuel; Vlugt, Thijs; Calero, Sofía; López, Núria (2012). "On the mechanism behind the instability of isoreticular metal-organic frameworks (IRMOFs) in humid environments". Chemistry: A European Journal. 18 (39): 12260–12266. doi:10.1002/chem.201201212.
- ↑ Stern, Abraham; Belof, Jonathan; Space, Brian; Eddaoudi, Mohamed (2012). "Understanding hydrogen sorption in a polar metal-organic framework with constricted channels". Journal of Chemical Physics. 136: 034705–034709. doi:10.1063/1.3668138.
- ↑ Dolgonos, Grygoriy (2005). "How many hydrogen molecules can be inserted into C60? Comments on the paper 'AM1 treatment of endohedrally hydrogen doped fullerene, nH2@C60' by L. Türker and S. Erkoç'". Journal of Molecular Structure: THEOCHEM. 723 (1–3): 239–241. doi:10.1016/j.theochem.2005.02.017.
- ↑ Tsao, Cheng-Si; Yu, MingSheng; Wang, Cheng-Yu; Liao, Pin-Yen; Chen, Hsin-Lung; Jeng, U-Ser; Tzeng, Yi-Ren; Chung, Tsui-Yun; Wu, Hsiu-Chu (2009). "Nanostructure and hydrogen spillover of bridged metal-organic frameworks". Journal of the American Chemical Society. 131 (4): 1404–1406. doi:10.1021/ja802741b. PMID 19140765.
- ↑ Mulfort, Karen L.; Farha, Omar K.; Stern, Charlotte L.; Sarjeant, Amy A.; Hupp, Joseph T. (2009). "Post-synthesis alkoxide formation within metal-organic framework materials: A strategy for incorporating highly coordinatively unsaturated metal ions". Journal of the American Chemical Society. 131 (11): 3866–3868. doi:10.1021/ja809954r. PMID 19292487.
- ↑ Huang, B. L.; Ni, Z.; Millward, A.; McGaughey, A. J. H.; Uher, C.; Kaviany, M.; Yaghi, O. (2007). "Thermal conductivity of a metal-organic framework (MOF-5): Part II. Measurement". International Journal of Heat and Mass Transfer. 50 (3–4): 405–411. doi:10.1016/j.ijheatmasstransfer.2006.10.001.
- ↑ McQuarrie, D. A.; Simon, J. D. (1997). Physical Chemistry: A Molecular Approach. Sausalito, CA: University Science Books.
- ↑ Belof, Jonathan; Stern, Abraham; Eddaoudi, Mohamed; Space, Brian (2007). "On the mechanism of hydrogen storage in a metal-organic framework material". Journal of the American Chemical Society. 129 (49): 15202–15210. doi:10.1021/ja0737164.
- ↑ Belof, Jonathan; Stern, Abraham; Space, Brian (2009). "A predictive model of hydrogen sorption for metal-organic materials". Journal of Physical Chemistry C. 113 (21): 9316–9320. doi:10.1021/jp901988e.
- 1 2 3 Zhao, Dan; Yan, Daqiang; Zhou, Hong-Cai (2008). "The current status of hydrogen storage in metal–organic frameworks". Energy & Environmental Science. 1 (1): 225–235. doi:10.1039/b808322n.
- ↑ Furukawa, Hiroyasu; Miller, Michael A.; Yaghi, Omar M. (2007). "Independent verification of the saturation hydrogen uptake in MOF-177 and establishment of a benchmark for hydrogen adsorption in metal–organic frameworks". Journal of Materials Chemistry. 17 (30): 3197–3204. doi:10.1039/b703608f.
- ↑ Lowell, S.; Shields, Joan E.; Thomas, Martin A.; Thommes, Matthias (2004). Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density. Springer Netherlands. doi:10.1007/978-1-4020-2303-3. ISBN 978-90-481-6633-6.
- 1 2 Züttel, Andreas (2004). "Hydrogen storage methods". Naturwissenschaften. 91 (4): 157–172. Bibcode:2004NW.....91..157Z. doi:10.1007/s00114-004-0516-x. PMID 15085273.
- ↑ Mitch Jacoby "Materials Chemistry: Metal-Organic Frameworks Go Commercial" Chemical & Engineering News, 91(51), December 23, 2013
- ↑ Jiri Cejka; Avelino Corma; Stacey Zones (27 May 2010). Zeolites and Catalysis: Synthesis, Reactions and Applications. John Wiley & Sons. ISBN 978-3-527-63030-1.
- ↑ Fujita, Makoto; Kwon, Yoon Jung; Washizu, Satoru; Ogura, Katsuyuki (1994). "Metal-Organic Frameworks: A Rapidly Growing Class of Versatile Nanoporous Material". Journal of the American Chemical Society. 116 (3): 1151. doi:10.1021/ja00082a055.
- ↑ Llabresixamena, F; Abad, A; Corma, A; Garcia, H (2007). "MOFs as catalysts: Activity, reusability and shape-selectivity of a Pd-containing MOF". Journal of Catalysis. 250 (2): 294. doi:10.1016/j.jcat.2007.06.004.
- ↑ Ravon, Ugo; Domine, Marcelo E.; Gaudillère, Cyril; Desmartin-Chomel, Arnold; Farrusseng, David (2008). "MOFs as acid catalysts with shape selectivity properties". New Journal of Chemistry. 32 (6): 937. doi:10.1039/B803953B.
- ↑ Chui, S. S.; Lo, Samuel M.-F.; Charmant, Jonathan P. H.; Orpen, A. Guy; Williams, Ian D. (1999). "A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n". Science. 283 (5405): 1148. Bibcode:1999Sci...283.1148C. doi:10.1126/science.283.5405.1148.
- ↑ Alaerts, Luc; Séguin, Etienne; Poelman, Hilde; Thibault-Starzyk, Frédéric; Jacobs, Pierre A.; De Vos, Dirk E. (2006). "Probing the Lewis Acidity and Catalytic Activity of the Metal–Organic Framework [Cu3(btc)2] (BTC=Benzene-1,3,5-tricarboxylate)". Chemistry: A European Journal. 12 (28): 7353. doi:10.1002/chem.200600220.
- ↑ Henschel, Antje; Gedrich, Kristina; Kraehnert, Ralph; Kaskel, Stefan (2008). "Catalytic properties of MIL-101". Chemical Communications (35): 4192. doi:10.1039/B718371B.
- ↑ Horike, Satoshi; Dincǎ, Mircea; Tamaki, Kentaro; Long, Jeffrey R. (2008). "Size-Selective Lewis-Acid Catalysis in a Microporous Metal-Organic Framework with Exposed Mn2+ Coordination Sites". Journal of the American Chemical Society. 130 (18): 5854. doi:10.1021/ja800669j.
- ↑ Chen, Long; Yang, Yong; Jiang, Donglin (2010). "CMPs as Scaffolds for Constructing Porous Catalytic Frameworks: A Built-in Heterogeneous Catalyst with High Activity and Selectivity Based on Nanoporous Metalloporphyrin Polymers". Journal of the American Chemical Society. 237 (26): 86. doi:10.1021/ja1028556.
- ↑ Rahiman, A. Kalilur; Rajesh, K.; Bharathi, K. Shanmuga; Sreedaran, S.; Narayanan, V. (2009). "Catalytic oxidation of alkenes by manganese(III) porphyrin-encapsulated Al, V, Si-mesoporous molecular sieves". Inorganica Chimica Acta. 352 (5): 1491. doi:10.1016/j.ica.2008.07.011.
- ↑ Mansuy, D.; Bartoli, J-F.; Momenteau, M. (1982). "Alkane hydroxylation catalyzed by metalloporhyrins : evidence for different active oxygen species with alkylhydroperoxides and iodosobenzene as oxidants". Tetrahedron Letters. 23 (27): 2781. doi:10.1016/S0040-4039(00)87457-2.
- ↑ Ingleson, Michael J.; Barrio, Jorge Perez; Bacsa, John; Dickinson, Calum; Park, Hyunsoo; Rosseinsky, Matthew J. (2008). "Generation of a solid Brønsted acid site in a chiral framework". Chemical Communications (11): 1287. doi:10.1039/B718443C.
- ↑ Hasegawa, Shinpei; Horike, Satoshi; Matsuda, Ryotaro; Furukawa, Shuhei; Mochizuki, Katsunori; Kinoshita, Yoshinori; Kitagawa, Susumu (2007). "Three-Dimensional Porous Coordination Polymer Functionalized with Amide Groups Based on Tridentate Ligand: Selective Sorption and Catalysis". Journal of the American Chemical Society. 129 (9): 2607–14. doi:10.1021/ja067374y. PMID 17288419.
- ↑ Hwang, Young Kyu; Hong, Do-Young; Chang, Jong-San; Jhung, Sung Hwa; Seo, You-Kyong; Kim, Jinheung; Vimont, Alexandre; Daturi, Marco; Serre, Christian; Férey, Gérard (2008). "Amine Grafting on Coordinatively Unsaturated Metal Centers of MOFs: Consequences for Catalysis and Metal Encapsulation". Angewandte Chemie International Edition. 47 (22): 4144. doi:10.1002/anie.200705998.
- ↑ Kim, Kimoon; Seo, Jung Soo; Whang, Dongmok; Lee, Hyoyoung; Jun, Sung Im; Oh, Jinho; Jeon, Young Jin (2000). "A homochiral metal–organic porous material for enantioselective separation and catalysis". Nature. 404 (6781): 982. doi:10.1038/35010088.
- ↑ Ohmori, O.; Fujita, M. (2004). "Heterogeneous catalysis of a coordination network: cyanosilylation of imines catalyzed by a Cd(II)-(4,4′-bipyridine) square grid complex". Chemical Communications (14): 1586. doi:10.1039/B406114B.
- ↑ Schlichte, Klaus; Kratzke, Tobias; Kaskel, Stefan (2004). "Improved synthesis, thermal stability and catalytic properties of the metal-organic framework compound Cu3(BTC)2". Microporous and Mesoporous Materials. 73: 81–85. doi:10.1016/j.micromeso.2003.12.027.
- ↑ Chui, S. S.; Lo, Samuel M.-F.; Charmant, Jonathan P. H.; Orpen, A. Guy; Williams, Ian D. (1999). "A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n". Science. 283 (5405): 1148. Bibcode:1999Sci...283.1148C. doi:10.1126/science.283.5405.1148.
- ↑ Horike, Satoshi; Dincǎ, Mircea; Tamaki, Kentaro; Long, Jeffrey R. (2008). "Size-Selective Lewis Acid Catalysis in a Microporous Metal-Organic Framework with Exposed Mn2+ Coordination Site". Journal of the American Chemical Society. 130 (18): 5854. doi:10.1021/ja800669j.
- ↑ Evans, Owen R.; Ngo, Helen L.; Lin, Wenbin (2001). "Chiral Porous Solids Based on Lamellar Lanthanide Phosphonates". Journal of the American Chemical Society. 123 (42): 10395. doi:10.1021/ja0163772.
- ↑ Kato, C; Hasegawa, M; Sato, T; Yoshizawa, A; Inoue, T; Mori, W (2005). "Microporous dinuclear copper(II) trans-1,4-cyclohexanedicarboxylate: heterogeneous oxidation catalysis with hydrogen peroxide and X-ray powder structure of peroxo copper(II) intermediate". Journal of Catalysis. 230: 226. doi:10.1016/j.jcat.2004.11.032.
- ↑ Han, Jong Woo; Hill, Craig L. (2007). "A Coordination Network That Catalyzes O2-Based Oxidations". Journal of the American Chemical Society. 129 (49): 15094. doi:10.1021/ja069319v.
- ↑ Hasegawa, Shinpei; Horike, Satoshi; Matsuda, Ryotaro; Furukawa, Shuhei; Mochizuki, Katsunori; Kinoshita, Yoshinori; Kitagawa, Susumu (2007). "Three-Dimensional Porous Coordination Polymer Functionalized with Amide Groups Based on Tridentate Ligand: Selective Sorption and Catalysis". Journal of the American Chemical Society. 129 (9): 2607–2614. doi:10.1021/ja067374y. PMID 17288419.
- ↑ Ferey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F.; Dutour, J.; Surblé, S.; Margiolaki, I. (2005). "A Chromium Terephthalate-Based Solid with Unusually Large Pore Volumes and Surface Area". Science. 309 (5743): 2040. Bibcode:2005Sci...309.2040F. doi:10.1126/science.1116275.
- ↑ Hwang, Young Kyu; Hong, Do-Young; Chang, Jong-San; Jhung, Sung Hwa; Seo, You-Kyong; Kim, Jinheung; Vimont, Alexandre; Daturi, Marco; Serre, Christian; Férey, Gérard (2008). "Amine Grafting on Coordinatively Unsaturated Metal Centers of MOFs: Conse3quences for Catalysis and Metal Encapsulation". Angewandte Chemie International Edition. 47 (22): 4144–4148. doi:10.1002/anie.200705998.
- ↑ Schröder, Felicitas; Esken, Daniel; Cokoja, Mirza; Van Den Berg, Maurits W. E.; Lebedev, Oleg I.; Van Tendeloo, Gustaaf; Walaszek, Bernadeta; Buntkowsky, Gerd; Limbach, Hans-Heinrich; Chaudret, Bruno; Fischer, Roland A. (2008). "Ruthenium Nanoparticles inside Porous [Zn4O(BDC)3] by hydrogenolysis of Adsorbed [Ru(cod)(cot)]: A Solid-State Reference System for Surfactant-Stabilized Ruthenium Colloids". Journal of the American Chemical Society. 130 (19): 6119–6130. doi:10.1021/ja078231u. PMID 18402452.
- ↑ Pan, L.; Liu, H.; Lei, X.; Haung, X.; Olson, D.; Turro, N.; Li, J. (2003). "A Recyclable Nanoporous Material Suitable for Ship-In-Bottle Synthesis and Large Hydrocarbon Sorption". Angewandte Chemie International Edition. 42 (5): 542–546. doi:10.1002/anie.200390156.
- ↑ Uemura, T.; Kitaura, R.; Ohta, Y.; Nagaoka, M.; Kitagawa, S. (2006). "Nanochannel-Promoted Polymerization of Substituted Acetylenes in Porous Coordination Polymers". Angewandte Chemie International Edition. 45 (25): 4112–4116. doi:10.1002/anie.200600333.
- ↑ Uemura, T.; Hiramatsu, D.; Kubota, Y.; Takata, M.; Kitagawa, S. (2007). "Topotactic Linear Radical Polymerization of Divinylbenzenes in Porous Coordination Polymers". Angewandte Chemie International Edition. 46 (26): 4987–4990. doi:10.1002/anie.200700242.
- ↑ Ezuhara, Takayoshi; Endo, Ken; Aoyama, Yasuhiro (1999). "Helical Coordination Polymers from Achiral Components in Crystals. Homochiral Crystallization, Homochiral Helix Winding in the Solid State, and Chirality Control by Seeding". Journal of the American Chemical Society. 121 (14): 3279. doi:10.1021/ja9819918.
- ↑ Wu, Shu-Ting; Wu, Yan-Rong; Kang, Qing-Qing; Zhang, Hui; Long, La-Sheng; Zheng, Zhiping; Huang, Rong-Bin; Zheng, Lan-Sun (2007). "Chiral Symmetry Breaking by Chemically Manipulating Statistical Fluctuation in Crystallization". Angewandte Chemie International Edition. 46 (44): 8475. doi:10.1002/anie.200703443.
- ↑ Kepert, C. J.; Prior, T. J.; Rosseinsky, M. J. (2000). "A Versatile Family of Interconvertible Microporous Chiral Molecular Frameworks: The First Example of Ligand Control of Network Chirality". Journal of the American Chemical Society. 122 (21): 5158. doi:10.1021/ja993814s.
- ↑ Bradshaw, Darren; Prior, Timothy J.; Cussen, Edmund J.; Claridge, John B.; Rosseinsky, Matthew J. (2004). "Permanent Microporosity and Enantioselective Sorption in a Chiral Open Framework". Journal of the American Chemical Society. 126 (19): 6106. doi:10.1021/ja0316420.
- ↑ Lin, Zhuojia; Slawin, Alexandra M. Z.; Morris, Russell E. (2007). "Chiral Induction in the Ionothermal Synthesis of a 3-D Coordination Polymer". Journal of the American Chemical Society. 129 (16): 4880. doi:10.1021/ja070671y.
- ↑ Hu, A.; Ngo, H.; Lin, W. (2004). "Remarkable 4,4′-Substituent Effects on Binap: Highly Enantioselective Ru Catalysts for Asymmetric Hydrogenation of β-Aryl Ketoesters and Their Immobilization in Room-Temperature Ionic Liquids". Angewandte Chemie International Edition. 43 (19): 2501–2504. doi:10.1002/anie.200353415.
- ↑ Wu, C.; Hu., A.; Zhang, L.; Lin, W. (June 2005). "A Homochiral Porous Metal-Organic Framework for Highly Enantioselective Heterogeneous Asymmetric Catalysis". Journal of the American Chemical Society. 127 (25): 8940–8941. doi:10.1021/ja052431t. PMID 15969557.
- ↑ Wu, Chuan-De; Lin, Wenbin (2007). "Heterogeneous Asymmetric Catalysis with Homochiral Metal–Organic Frameworks: Network-Structure-Dependent Catalytic Activity". Angewandte Chemie International Edition. 46 (7): 1075. doi:10.1002/anie.200602099.
- ↑ Cho, So-Hye; Ma, Baoqing; Nguyen, Sonbinh T.; Hupp, Joseph T.; Albrecht-Schmitt, Thomas E. (2007). "A Metal-Organic Framework Material That Functions as an Enantioselective Catalyst for Olefin Epoxidation". Chemical Communications (24): 2563. doi:10.1039/b600408c.
- ↑ Hu, Aiguo; Ngo, Helen L.; Lin, Wenbin (2003). "Chiral Porous Hybrid Solids for Practical Heterogeneous Asymmetric Hydrogenation of Aromatic Ketones". Journal of the American Chemical Society. 125 (38): 11490–1. doi:10.1021/ja0348344. PMID 13129339.
- ↑ Farrusseng, David; Aguado, Sonia; Pinel, Catherine (2009). "Metal–Organic Frameworks: Opportunities for Catalysis". Angewandte Chemie International Edition. 48 (41): 7502–7513. doi:10.1002/anie.200806063.
- ↑ Eddaoudi, M.; Kim, Jaheon; Wachter, J. B.; Chae, H. K.; O'Keeffe, M.; Yaghi, O. M. (2001). "Porous Metal-Organic Polyhedra: 25 Å Cuboctahedron Constructed from 12 Cu2(CO2)4 Paddle-Wheel Building Blocks". Journal of the American Chemical Society. 123 (18): 4368. doi:10.1021/ja0104352.
- ↑ Furukawa, Hiroyasu; Kim, Jaheon; Ockwig, Nathan W.; o’Keeffe, Michael; Yaghi, Omar M. (2008). "Control of Vertex Geometry, Structure Dimensionality, Functionality, and Pore Metrics in the Reticular Synthesis of Crystalline Metal-Organic Frameworks and Polyhedra". Journal of the American Chemical Society. 130 (35): 11650. doi:10.1021/ja803783c.
- ↑ Surblé, Suzy; Serre, Christian; Mellot-Draznieks, Caroline; Millange, Franck; Férey, Gérard (2006). "A new isoreticular class of metal-organic-frameworks with the MIL-88 topology". Chemical Communications (3): 284. doi:10.1039/B512169H.
- ↑ Sudik, Andrea C.; Millward, Andrew R.; Ockwig, Nathan W.; Côté, Adrien P.; Kim, Jaheon; Yaghi, Omar M. (2005). "Design, Synthesis, Structure, and Gas (N2, Ar, CO2, CH4, and H2) Sorption Properties of Porous Metal-Organic Tetrahedral and Heterocuboidal Polyhedra". Journal of the American Chemical Society. 127 (19): 7110. doi:10.1021/ja042802q.
- ↑ Degnan, T (2003). "The implications of the fundamentals of shape selectivity for the development of catalysts for the petroleum and petrochemical industries". Journal of Catalysis. 216: 32. doi:10.1016/S0021-9517(02)00105-7.
- ↑ Kuc, A.; Enyashin, A.; Seifert, G. (2007). "Metal-Organic Frameworks: Structural, Energetic, Electronic, and Mechanical Properties". The Journal of Physical Chemistry B. 111 (28): 8179. doi:10.1021/jp072085x.
- ↑ Esswein, Arthur J.; Nocera, Daniel G. (2007). "Hydrogen Production by Molecular Photocatalysis". Chemical Reviews. 107 (10): 4022. doi:10.1021/cr050193e.
- ↑ Yang, Chi; Messerschmidt, Marc; Coppens, Philip; Omary, Mohammad A. (2006). "Trinuclear Gold(I) Triazolates: A New Class of Wide-Band Phosphors and Sensors". Inorganic Chemistry. 45 (17): 6592. doi:10.1021/ic060943i.
- ↑ Fuentes-Cabrera, Miguel; Nicholson, Donald M.; Sumpter, Bobby G.; Widom, Mike (2005). "Electronic structure and properties of isoreticular metal-organic frameworks: The case of M-IRMOF1 (M=Zn, Cd, Be, Mg, and Ca)". The Journal of Chemical Physics. 123 (12): 124713. Bibcode:2005JChPh.123l4713F. doi:10.1063/1.2037587.
- ↑ Gascon, Jorge; Hernández-Alonso, María D.; Almeida, Ana Rita; Van Klink, Gerard P. M.; Kapteijn, Freek; Mul, Guido (2008). "Isoreticular MOFs as Efficient Photocatalysts with Tunable Band Gap: An Operando FTIR Study of the Photoinduced Oxidation of Propylene". ChemSusChem. 1 (12): 981. doi:10.1002/cssc.200800203.
- ↑ Horcajada, Patricia; Chalati, Tamim; Serre, Christian; Gillet, Brigitte; Sebrie, Catherine; Baati, Tarek; Eubank, Jarrod F.; Heurtaux, Daniela; Clayette, Pascal. "Porous metal–organic-framework nanoscale carriers as a potential platform for drug delivery and imaging". Nature Materials. 9 (2): 172–178. doi:10.1038/nmat2608.
- ↑ Mendoza-Cortés, José L.; Han, Sang Soo; Furukawa, Hiroyasu; Yaghi, Omar M.; Goddard Wa, William A. (2010). "Adsorption Mechanism and Uptake of Methane in Covalent Organic Frameworks: Theory and Experiment". The Journal of Physical Chemistry A. 114 (40): 10824–33. doi:10.1021/jp1044139. PMID 20845983.
- 1 2 Mendoza-Cortes, Jose L.; Pascal, Tod A.; Goddard Wa, William A. (2011). "Design of Covalent Organic Frameworks for Methane Storage". The Journal of Physical Chemistry A. 115 (47): 13852–7. doi:10.1021/jp209541e. PMID 21992457.
- ↑ Markoff, John (January 9, 2014). "Designing the Next Wave of Computer Chips". New York Times. Retrieved January 10, 2014.
- ↑ Talin, A. A.; Centrone, A.; Ford, A. C.; Foster, M. E.; Stavila, V.; Haney, P.; Kinney, R. A.; Szalai, V.; El Gabaly, F.; Yoon, H. P.; Léonard, F.; Allendorf, M. D. (2014). "Tunable Electrical Conductivity in Metal-Organic Framework Thin-Film Devices". Science. 343 (6166): 66–69. doi:10.1126/science.1246738. PMID 24310609.
- ↑
- ↑ Sheberla, D.; Sun, L.; Blood-Forsythe, M. A.; Er, S. L.; Wade, C. R.; Brozek, C. K.; Aspuru-Guzik, A. N.; Dincă, M. (2014). "High Electrical Conductivity in Ni3(2,3,6,7,10,11-hexaiminotriphenylene)2, a Semiconducting Metal–Organic Graphene Analogue". Journal of the American Chemical Society: 140425154202006. doi:10.1021/ja502765n.
- ↑ Liang, Kang (2015). "Biomimetic mineralization of metal-organic frameworks as protective coatings for biomacromolecules". Nature Communications. 6. doi:10.1038/ncomms8240.
- ↑ Jamali, Abbas (2016). "Lanthanide metal–organic frameworks as selective microporous materials for adsorption of heavy metal ions". Dalton Transactions. 45 (22): 9193–9200. doi:10.1039/C6DT00782A.
- ↑ Choi, Sunho; Drese, JeffreyH.; Jones, ChristopherW. (2009-09-21). "Adsorbent Materials for Carbon Dioxide Capture from Large Anthropogenic Point Sources". ChemSusChem. 2 (9): 796–854. doi:10.1002/cssc.200900036. ISSN 1864-564X.
- 1 2 3 Sumida, Kenji; Rogow, David L.; Mason, Jarad A.; McDonald, Thomas M.; Bloch, Eric D.; Herm, Zoey R.; Bae, Tae-Hyun; Long, Jeffrey R. (2012-02-08). "Carbon Dioxide Capture in Metal–Organic Frameworks". Chemical Reviews. 112 (2): 724–781. doi:10.1021/cr2003272. ISSN 0009-2665.
- ↑ Berger, Adam Hughmanick; Bhown, Abhoyjit S. (2011-01-01). "Comparing physisorption and chemisorption solid sorbents for use separating CO2 from flue gas using temperature swing adsorption". Energy Procedia. 10th International Conference on Greenhouse Gas Control Technologies. 4: 562–567. doi:10.1016/j.egypro.2011.01.089.
- ↑ Smit, Berend; Reimer, Jeffrey A.; Oldenburg, Curtis M.; Bourg, Ian C. (2014). Introduction to Carbon Capture and Sequestration. London: Imperial College Press. ISBN 978-1-78326-328-8.
- ↑ Millward, Andrew R.; Yaghi, Omar M. (2005-12-01). "Metal−Organic Frameworks with Exceptionally High Capacity for Storage of Carbon Dioxide at Room Temperature". Journal of the American Chemical Society. 127 (51): 17998–17999. doi:10.1021/ja0570032. ISSN 0002-7863.
- ↑ Lesch, David A. "Carbon Dioxide Removal from Flue Gas Using Microporous Metal Organic Frameworks". doi:10.2172/1003992.