Non-covalent interactions
A non-covalent interaction differs from a covalent bond in that it does not involve the sharing of electrons, but rather involves more dispersed variations of electromagnetic interactions between molecules or within a molecule.[1] The chemical energy released in the formation of non-covalent interactions is typically on the order of 1-5 kcal/mol (1000–5000 calories per 6.02 x 10^23 molecules).[2] Non-covalent interactions can be classified into different categories, such as electrostatic, π-effects, van der Waals forces, and hydrophobic effects.[1][2]
Non-covalent interactions [3] are critical in maintaining the three-dimensional structure of large molecules, such as proteins and nucleic acids. In addition, they are also involved in many biological processes in which large molecules bind specifically but transiently to one another (see the properties section of the DNA page). These interactions also heavily influence drug design, crystallinity and design of materials, particularly for self-assembly, and, in general, the synthesis of many organic molecules.[1][4][5][6]
Intermolecular forces are non-covalent interactions that occur between different molecules, rather than between different atoms of the same molecule [1]
Electrostatic interactions
For a more detailed introduction to electrostatics, see Coulomb's Law
Ionic

Ionic interactions involve the attraction of ions or molecules with full permanent charges of opposite signs. For example, sodium fluoride involves the attraction of the positive charge on sodium (Na+) with the negative charge on fluoride (F−). These bonds are harder to break than covalent bonds because there is a strong electrostatic interaction between oppositely charged ions. However, this particular interaction is easily broken upon addition to water, or other highly polar solvents.
These interactions can also be seen in molecules with a localized charge on a particular atom. For example, the full negative charge associated with ethoxide, the conjugate base of ethanol, is most commonly accompanied by the positive charge of an alkali metal salt such as the sodium cation (Na+).
Hydrogen bonding

A hydrogen bond (H-bond), is a specific type of interaction that involves dipole-dipole attraction between a partially positive hydrogen atom and a highly electronegative, partially negative oxygen, nitrogen, sulfur, or fluorine atom (not covalently bound to said hydrogen atom). It is not a covalent bond, but instead is classified as a strong non-covalent interaction. It is responsible for why water is a liquid at room temperature and not a gas (given water's low molecular weight). Most commonly, the strength of hydrogen bonds lies between 0 - 4 kcal/mol, but can sometimes be as strong as 40 kcal/mol[1]
Halogen bonding

Halogen bonding is a type of non-covalent interaction which does not involve the formation nor breaking of actual bonds, but rather is similar to the dipole-dipole interaction known as hydrogen bonding. In halogen bonding, a halogen atom acts as an electrophile, or electron-seeking species, and forms a weak electrostatic interaction with a nucleophile, or electron-rich species. The nucleophilic agent in these interactions tends to be highly electronegative (such as oxygen, nitrogen, or sulfur), or may be anionic, bearing a negative formal charge. As compared to hydrogen bonding, the halogen atom takes the place of the partially positively charged hydrogen as the electrophile.
Halogen bonding should not be confused with halogen-aromatic interactions, as the two are related but differ by definition. Halogen-aromatic interactions involve an electron-rich aromatic π-cloud as a nucleophile; halogen bonding is restricted to monatomic nucleophiles.[4]
Van der Waals forces
Van der Waals Forces are a subset of electrostatic interactions involving permanent or induced dipoles (or multipoles). These include the following:
- permanent dipole-dipole interactions, alternatively called the Keesom force
- dipole-induced dipole interactions, or the Debye force
- induced dipole-induced dipole interactions, commonly referred to as London dispersion forces
Hydrogen bonding and halogen bonding are typically not classified as Van der Waals forces.
Dipole-dipole

Dipole-dipole interactions are electrostatic interactions between permanent dipoles in molecules. These interactions tend to align the molecules to increase attraction (reducing potential energy). Normally, dipoles are associated with electronegative atoms, including oxygen, nitrogen, sulfur, and fluorine.
For example, acetone, the active ingredient in some nail polish removers, has a net dipole associated with the carbonyl (see figure 2). Since oxygen is more electronegative than the carbon that is covalently bonded to it, the electrons associated with that bond will be closer to the oxygen than the carbon, creating a partial negative charge (δ−) on the oxygen, and a partial positive charge (δ+) on the carbon. They are not full charges because the electrons are still shared through a covalent bond between the oxygen and carbon. If the electrons were no longer being shared, then the oxygen-carbon bond would be an electrostatic interaction.

Often molecules contain dipolar groups, but have no overall dipole moment. This occurs if there is symmetry within the molecule that causes the dipoles to cancel each other out. This occurs in molecules such as tetrachloromethane. Note that the dipole-dipole interaction between two individual atoms is usually zero, since atoms rarely carry a permanent dipole. See atomic dipoles.
Dipole-induced dipole
A dipole-induced dipole interaction (Debye force) is due to the approach of a molecule with a permanent dipole to another non-polar molecule with no permanent dipole. This approach causes the electrons of the non-polar molecule to be polarized toward or away from the dipole (or "induce" a dipole) of the approaching molecule.[7] Specifically, the dipole can cause electrostatic attraction or repulsion of the electrons from the non-polar molecule, depending on orientation of the incoming dipole. [7] Atoms with larger atomic radii are considered more "polarizable" and therefore experience greater attraction as a result of the Debye force.
London dispersion forces
London dispersion forces are the weakest type of non-covalent interaction. They are also known as "induced dipole-induced dipole interactions" and present between all molecules, even those which inherently do not have permanent dipoles. They are caused by the temporary repulsion of electrons away from the electrons of a neighboring molecule, leading to a partially positive dipole on one molecule and a partially negative dipole on another molecule.[5] Hexane is a good example of a molecule with no polarity or highly electronegative atoms, yet is a liquid at room temperature due mainly to London dispersion forces. In this example, when one hexane molecule approaches another, a temporary, weak partially negative dipole on the incoming hexane can polarize the electron cloud of another, causing a partially positive dipole on that hexane molecule. While these interactions are short-lived and very weak, they can be responsible for why certain non-polar molecules are liquids at room temperature.
π-effects
π-effects can be broken down into numerous categories, including π-π interactions, cation-π & anion-π interactions, and polar-π interactions. In general, π-effects are associated with the interactions of molecules with the π-systems of conjugated molecules such as benzene.[1]
π-π interaction
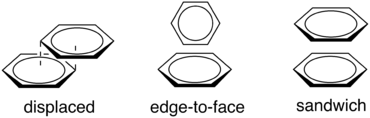
π-π interactions are associated with the interaction between the π-orbitals of a molecular system.[1] For a simple example, a benzene ring, with its fully conjugated π cloud, will interact in two major ways (and one minor way) with a neighboring benzene ring through a π-π interaction (see figure 3). The two major ways that benzene stacks are edge-to-face, with an enthalpy of ~2 kcal/mol, and displaced (or slip stacked), with an enthalpy of ~2.3 kcal/mol.[1] Interestingly, the sandwich configuration is not nearly as stable of an interaction as the previously two mentioned due to high electrostatic repulsion of the electrons in the π orbitals.[1]
Cation-π and anion-π interaction
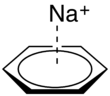
Cation-π interactions involve the positive charge of a cation interacting with the electrons in a π-system of a molecule.[1] This interaction is surprisingly strong (as strong or stronger than H-bonding in some contexts),[1] and has many potential applications in chemical sensors.[8] For example, the sodium ion can easily sit atop the π cloud of a benzene molecule, with C6 symmetry (for more on point groups and molecular symmetry, see the Wikipedia page on point groups) (See figure 4).
Anion-π interactions are very similar to cation-π interactions, but reversed. In this case, an anion sits atop an electron-poor π-system, usually established by the placement of electron-withdrawing substituents on the conjugated molecule[9]

Polar-π
Polar-π interactions involve molecules with permanent dipoles (such as water) interacting with the quadrupole moment of a π-system (such as that in benzene (see figure 5). While not as strong as a cation-π interaction, these interactions can be quite strong (~1-2 kcal/mol), and are commonly involved in protein folding and crystallinity of solids containing both hydrogen bonding and π-systems.[1] In fact, any molecule with a hydrogen bond donor (hydrogen bound to a highly electronegative atom) will have favorable electrostatic interactions with the electron-rich π-system of a conjugated molecule.
Hydrophobic effect
The hydrophobic effect is the desire for non-polar molecules to aggregate in aqueous solutions in order to separate from water.[10] This phenomenon leads to minimum exposed surface area of non-polar molecules to the polar water molecules (typically spherical droplets), and is commonly used in biochemistry to study protein folding and other various biological phenomenon.[10] The effect is also commonly seen when mixing various oils (including cooking oil) and water. Over time, oil sitting on top of water will begin to aggregate into large flattened spheres from smaller droplets, eventually leading to a film of all oil sitting atop a pool of water.
Examples
Drug design
Most pharmaceutical drugs are small molecules which elicit a physiological response by "binding" to enzymes or receptors, causing an increase or decrease in the enzyme's ability to function. The binding of a small molecule to a protein is governed by a combination of steric, or spatial considerations, in addition to various non-covalent interactions, although some drugs do covalently modify an active site (see irreversible inhibitors). Using the "lock and key model" of enzyme binding, a drug (key) must be of roughly the proper dimensions to fit the enzyme's binding site (lock).[11] Using the appropriately sized molecular scaffold, drugs must also interact with the enzyme non-covalently in order to maximize binding affinity binding constant and reduce the ability of the drug to dissociate from the binding site. This is achieved by forming various non-covalent interactions between the small molecule and amino acids in the binding site, including: hydrogen bonding, electrostatic interactions, pi stacking, van der Waals interactions, and dipole-dipole interactions.
Protein folding and structure

The folding of most proteins from a primary (linear) sequence of amino acids to a three-dimensional structure is governed by many factors, including non-covalent interactions. The first ~5 milliseconds of folding are primarily dependent on van der Waals forces, whereby the protein folds so as to orient nonpolar amino acids in the interior of the globular protein, while more polar amino acid residues are exposed to aqueous solvent. This phase is known as the hydrophobic collapse, when nonpolar non-covalent interactions exclude water from the interior of the developing 3D protein structure.
After this initial "burst phase," more polar non-covalent interactions take over. Between 5 and 1000 milliseconds after protein folding initiation, three-dimensional structures of proteins, known as secondary and tertiary structures, are stabilized by formation of hydrogen bonds, in addition to disulfide bridges (covalent linkages). Through a series of small conformational changes, spatial orientations are modified so as to arrive at the most energetically minimized orientation achievable. The folding of proteins is often facilitated by enzymes known as molecular chaperones[12] Sterics, bond strain, and angle strain also play major roles in the folding of a protein from its primary sequence to its tertiary structure.
Single tertiary protein structures can also assemble to form protein complexes composed of multiple independently folded subunits. As a whole, this is called a protein's quaternary structure. The quaternary structure is generated by the formation of relatively strong non-covalent interactions, such as hydrogen bonds, between different subunits to generate a functional polymeric enzyme.[13] Some proteins also utilize non-covalent interactions to bind cofactors in the active site during catalysis, however a cofactor can also be covalently attached to an enzyme. Cofactors can be either organic or inorganic molecules which assist in the catalytic mechanism of the active enzyme. The strength with which a cofactor is bound to an enzyme may vary greatly; non-covalently bound cofactors are typically anchored by hydrogen bonds or electrostatic interactions.
Boiling points
Non-covalent interactions have a significant effect on the boiling point of a liquid. Boiling point is defined as the temperature at which the vapor pressure of a liquid is equal to the pressure surrounding the liquid. More simply, it is the temperature at which a liquid becomes a gas. As one might expect, the stronger the non-covalent interactions present for a substance, the higher its boiling point. For example, consider three compounds of similar chemical composition: sodium n-butoxide (C4H9ONa), diethyl ether (C4H10O), and n-butanol (C4H9OH).
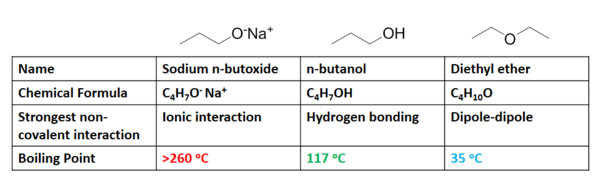
The predominant non-covalent interactions associated with each species in solution are listed in the above figure. As previously discussed, ionic interactions require considerably more energy to break than hydrogen bonds, which in turn are require more energy than dipole-dipole interactions. The trends observed in their boiling points (figure 8) shows exactly the correlation expected, where sodium n-butoxide requires significantly more heat energy (higher temperature) to boil than n-butanol, which boils at a much higher temperature than diethyl ether. The heat energy required for a compound to change from liquid to gas is associated with the energy required to break the intermolecular forces each molecule experiences in its liquid state.
References
- 1 2 3 4 5 6 7 8 9 10 11 12 Anslyn, Eric (2004). Modern Physical Organic Chemistry. Sausalito, CA: University Science. ISBN 978-1-891389-31-3.
- 1 2 Noncovalent bonds – Molecular Cell Biology (textbook), Lodish, Berk, Zipursky, Matsudaira, Baltimore, Darnell.
- ↑ Noncovalent bonding in Supramolecular Chemistry - Christoph A. Schalley
- 1 2 Cockroft, Scott L.; Hunter, Christopher A. (1 January 2007). "Chemical double-mutant cycles: dissecting non-covalent interactions". Chemical Society Reviews. 36 (2): 172. doi:10.1039/b603842p.
- 1 2 Brown, Theodore; et al. (2009). Chemistry : the central science (11th ed.). Upper Saddle River, NJ: Pearson Prentice Hall. ISBN 978-0-13-600617-6.
- ↑ Eisler, Matthew (2010). Encyclopedia of nanoscience and society. Thousand Oaks, Calif.: Sage. ISBN 978-1-4129-7209-3.
- 1 2 "Induced-Dipole Forces". Retrieved 11 November 2013.
- ↑ Sastry, G. N., & Mahadevi, A. S. (2013). Cation-π interaction: Its role and relevevance in chemistry, biology, and material science. Chemical Reviews, 113, 2100
- ↑ Quiñonero, David; Garau, Carolina; Rotger, Carmen; Frontera, Antonio; Ballester, Pablo; Costa, Antonio; Deyà, Pere M. (16 September 2002). "Anion–π Interactions: Do They Exist?". Angewandte Chemie International Edition. 41 (18): 3389–3392. doi:10.1002/1521-3773(20020916)41:18<3389::AID-ANIE3389>3.0.CO;2-S.
- 1 2 IUPAC. "Compendium of Chemical Terminology". Retrieved 11 November 2013.
- ↑ "Biomolecules: Enzymes". ChemPages Netorials. University of Wisconsin - Madison. Retrieved 27 October 2013.
- ↑ Voet, Donald., & Voet, Judith G. (2010). Biochemistry (4th ed.). Hoboken, NJ: John Wiley & Sons. ISBN 978-0-470-57095-1.
- ↑ Silverman, Richard B. (2004). The organic chemistry of drug design and drug action (2. ed.). Amsterdam [u.a.]: Elsevier. ISBN 978-0-12-643732-4.
Non-Covalent Interactions in the Synthesis and Design of New Compounds. A. M. Maharramov, K.T. Mahmudov, M.N. Kopylovich, A.J.L. Pombeiro, (Eds.), John Wiley & Sons, Inc., Hoboken, New Jersey, 2016.ISBN 978-1-119-10989-1