Photopolymer
A photopolymer is a polymer that changes its properties when exposed to light, often in the ultraviolet or visible region of the electromagnetic spectrum.[1] These changes are often manifested structurally, for example hardening of the material occurs as a result of cross-linking when exposed to light. An example is shown below depicting a mixture of monomers, oligomers, and photoinitiators that conform into a hardened polymeric material through a process called curing.[2][3] A wide variety of technologically useful applications rely on photopolymers, for example some enamels and varnishes depend on photopolymer formulation for proper hardening upon exposure to light. In some instances, an enamel can cure in a fraction of a second when exposed to light, as opposed to thermally cured enamels which can require half an hour or longer.[4] Curable materials are widely used for medical, printing, and photoresist technologies.
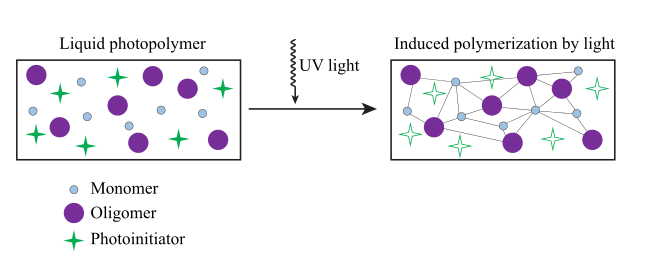
Changes in structural and chemical properties can be induced internally by chromophores that the polymer subunit already possesses, or externally by addition of photosensitive molecules. Typically a photopolymer consists of a mixture of multifunctional monomers and oligomers in order to achieve the desired physical properties, and therefore a wide variety of monomers and oligomers have been developed that can polymerize in the presence of light either through internal or external initiation. Photopolymers undergo a process called curing, where oligomers are cross-linked upon exposure to light, forming what is known as a network polymer. The result of photo curing is the formation of a thermoset network of polymers. One of the advantages of photo-curing is that it can be done selectively using high energy light sources, for example lasers, however, most systems are not readily activated by light, and in this case a photoinitiator is required. Photoinitiators are compounds that upon radiation of light decompose into reactive species that activate polymerization of specific functional groups on the oligomers.[5] An example of a mixture that undergoes cross-linking when exposed to light is shown below. The mixture consists of monomeric styrene and oligomeric acrylates.[6]

Most commonly, photopolymerized systems are typically cured through UV radiation, since ultraviolet light is more energetic; however, the development of dye-based photoinitiator systems have allowed for the use of visible light, having potential advantages of processes that are more simple and safe to handle.[7] UV curing in industrial processes has greatly expanded over the past several decades. Many traditional thermally cured and solvent-based technologies can be replaced by photopolymerization technologies. The advantages of photopolymerization over thermally cured polymerization include high rates of polymerization and environmental benefits from elimination of volatile organic solvents.[1]
There are two general routes for photoinitiation: free radical and ionic.[1][4] The general process involves doping a batch of neat polymer with small amounts of photoinitiator, followed by selective radiation of light, resulting a highly cross-linked product. Many of these reactions do not require solvent which eliminates termination path via reaction of initiators with solvent and impurities, in addition to decreasing the overall cost.[8]
Ionic mechanism
In ionic curing process, an ionic photoinitiator is used to activate the functional group of the oligomers that are going to participate in cross-linking. Typically photopolymerization is a very selective process and it is crucial that the polymerization takes place only where it is desired to do so. In order to satisfy this liquid neat oligomer can be doped with either anionic or cationic photoinitiators that will initiate polymerization only when radiated with light. Monomers, or functional groups, employed in cationic photopolymerization include: styrenic compounds, vinyl ethers, N-vinyl carbazoles, lactones, lactams, cyclic ethers, cyclic acetals, and cyclic siloxanes. The majority of ionic photoinitiators fall under the cationic class, anionic photoinitiators are considerably less investigated.[5] There are several classes of cationic initiators including: Onium salts, organometallic compounds and pyridinium salts.[5] As mentioned earlier, one of the drawbacks of the photoinitiators used for photopolymerization is that they tend to absorb in the short UV region.[7] Photosensitizers, or chromophores, that absorb in a much longer wavelength region can be employed to excite the photoinitiators through an energy transfer.[5] Other modifications to these types of systems are free radical assisted cationic polymerization. In this case, a free radical is formed from another specie in solution that reacts with the photoinitiator in order to start polymerization. Although there are a diverse group of compounds activated by cationic photoinitiators, the compounds that find most industrial uses contain epoxides, oxetanes, and vinyl ethers.[9] One of the advantages to using cationic photopolymerization is that once the polymerization has begun it is no longer sensitive to oxygen and does not require an inert atmosphere to perform well.[1]
- Photolysis
-
- M = Monomer
![{\displaystyle {\begin{matrix}{}\\{\ce {[R'-R+X^{-}]->[hv]{[R'-R+X-]^{\ast }}->{R^{.+}}+{R'^{.}}+X^{-}->[{\ce {MH}}]{{R+}-H}+{M-R'}+X^{-}->{R}+H+X^{-}}}\\{}\end{matrix}}}](../I/m/ff243582d9b8b2dcfb0ee43770265ae987934acd.svg "Photolysis of cationic photoinitiator")
Cationic photoinitiators
The proposed mechanism for cationic photopolymerization begins with the photoexcitation of the initiator. Once excited, both homolytic cleavage and dissociation of a counter anion takes place, generating cationic radical (R), an aryl radical(R') and unaltered counter anion (X). The abstraction of a lewis acid, in figure above a hydrogen, by the cationic radical produces a very weakly bound hydrogen and a free radical. The acid is further deprotonated by the anion(X) in solution generating a lewis acid with the starting anion (X) as a counter ion. It is thought that the acidic proton generated is what ultimately initiates the polymerization.[10]
Onium salts
Since their discovery in the 1970s aryl onium salts, more specifically iodonium and sulfonium salts, have received much attention and have found many industrial applications. Other, less common, onium salts not mentioned here include ammonium and phosphonium salts.[9]

The typical onium compound used as a photoinitiator contains two or three arene groups for iodonium and sulfonium respectively. Onium salts generally absorb short wavelength light in the UV region spanning from 225–300 nm.[11] One characteristic that is crucial to the performance of the onium photoinitiators is that the counter anion is non-nucleophilic. Since the Brønsted acid generated during the initiation step is considered the active initiator for polymerization, there is a termination route where the counter ion of the acid could act as the nucleophile instead of a functional groups on the oligomer. Common counter anions include: BF−
4, PF−
6, AsF−
6, SbF−
6. There is an indirect relationship between the size of the counter ion and percent conversion.
Organometallic
Although less common, transition metal complexes can act as cationic photoinitiators as well. In general, the mechanism is more simplistic than the onium ions previously described. Most photoinitiators of this class consist of a metal salt with a non-nucleophilic counter anion. For example, ferrocinium salts have received much attention for commercial applications.[12] The absorption band for ferrocinium salt derivatives are in a much longer, and sometimes visible, region. Upon radiation the metal center loses a ligand(s) and the ligand(s) is replaced by functional groups that begin the polymerization. One of the drawbacks of this method is a greater sensitivity to oxygen. There are also several organometallic anionic photoinitiators which react through a similar mechanism. For the anionic case, excitation of a metal center is followed by either heterolytic bond cleavage or electron transfer generating the active anionic initiator.[5]
Pyridinium salts
Generally pyridinium photoinitiators are N-substituted pyridine derivatives, with a positive charge placed on the nitrogen. The counter ion is in most cases a non-nucleophilic anion. Upon radiation, homolytic bond cleavage takes place generating a pyridinium cationic radical and a neutral free radical. In most cases, a hydrogen atom is abstracted from the oligomer by the pyridinium radical. The free radical generated from the hydrogen abstraction is then terminated by the free radical in solution. This results in a strong pyridinium acid that can initiate polymerization.[13]
Free radical mechanism
Before the free radical nature of certain polymerizations was determined, certain monomers were observed to polymerize when exposed to light. The first to demonstrate the photoinduced free radical chain reaction of vinyl bromide was Ivan Ostromislensky, a Russian chemist who also studied the polymerization of synthetic rubber. Subsequently many compounds were found to become dissociated by light and found immediate use as photoinitiators in the polymerization industry.[1] In the free radical mechanism of radiation curable systems light absorbed by a photoinitiator generates free-radicals which induce cross-linking reactions of a mixture of functionalized oligomers and monomers to generate the cured film [14] Photocurable materials that form through the free-radical mechanism undergo chain-growth polymerization, which includes three basic steps: initiation, chain propagation, and chain termination. The three steps are depicted in the scheme below, where R• represents the radical that forms upon interaction with radiation during initiation, and M is a monomer.[4] The active monomer that is formed is then propagated to create growing polymeric chain radicals. In photocurable materials the propagation step involves reactions of the chain radicals with reactive double bonds of the prepolymers or oligomers. The termination reaction usually proceeds through combination, in which two chain radicals are joined together, or through disproportionation, which occurs when an atom (typically hydrogen) is transferred from one radical chain to another resulting in two polymeric chains.
- Initiation
- Propagation
- Termination
- combination
- dispropotionation




Most composites that cure through radical chain growth contain a diverse mixture of oligomers and monomers with functionality that can range from 2-8 and molecular weights from 500-3000. In general, monomers with higher functionality result is a tighter crosslinking density of the finished material.[5] Typically these oligomers and monomers alone do not absorb sufficient energy for the commercial light sources used, therefore photoinitiators are included.,[4][14]
Photoinitiators
There are two types of free-radical photoinitators: A two component system where the radical is generated through abstraction of a hydrogen atom from a donor compound (also called co-initiator), and a one component system where two radicals are generated by cleavage. Examples of each type of free-radical photoinitiator is shown below.[14]

Benzophenone, Xanthones, and Quinones are examples of abstraction type photoinitiators, with common donor compounds being aliphatic amines. The resulting R• species from the donor compound becomes the initiator for the free radical polymerization process, while the radical resulting from the starting photoinitiator (benzophenone in the example shown above) is typically unreactive.
Benzoin ethers, Acetophenones, Benzoyl Oximes, and Acylphosphines are some examples of cleavage-type photoinitiators. Cleavage readily occurs for the species to give two radicals upon absorption of light, and both radicals generated can typically initiate polymerization. Cleavage type photoinitiators do not require a co-initiator, such as aliphatic amines. This can be beneficial since amines are also effective chain transfer species. Chain-transfer processes reduce the chain length and ultimately the crosslink density of the resulting film.
Oligomers and monomers
The properties of a photocured material, such as flexibility, adhesion, and chemical resistance are provided by the functionalized oligomers present in the photocurable composite. Oligomers are typically epoxides, urethanes, polyethers, or polyesters, each of which provide specific properties to the resulting material. Each of these oligomers are typically functionallized by an acrylate. An example shown below is an epoxy oligomer that has been functionalized by acrylic acid. Acrylated epoxies are useful as coatings on metallic substrates, and result in glossy hard coatings. Acrylated urethane oligomers are typically abrasion resistant, tough, and flexible making ideal coatings for floors, paper, printing plates, and packaging materials. Acrylated polyethers and polyesters result in very hard solvent resistant films, however, polyethers are prone to UV degradation and therefore are rarely used in UV curable material. Often formulations are composed of several types of oligomers to achieve the desirable properties for a material.[4]

The monomers used in radiation curable systems help control the speed of cure, crosslink density, final surface properties of the film, and viscosity of the resin. Examples of monomers include styrene, N-Vinylpyrrolidone, and acrylates. Styrene is a low cost monomer and provides a fast cure, N-vinylpyrrolidone results in a material that is highly flexible when cured, has low toxicity, and acrylates are highly reactive, allowing for rapid cure rates, and are highly versatile with monomer functionality ranging from monofunctional to tetrafunctional. Like oligomers, several types of monomers can be employed to achieve the desirable properties of the final material.[4]
Applications
Photopolymerization is a widely used technology, used in applications ranging from imaging to biomedical uses. Below is a description of just some photopolymerization applications.
Medical uses
Dentistry is one market where free radical photopolymers have found wide usage as adhesives, sealant composites, and protective coatings. These dental composites are based on a camphorquinone photoinitiator and a matrix containing methacrylate oligomers with inorganic fillers such as silicon dioxide. Photocurable adhesives are also used in the production of catheters, hearing aids, surgical masks, medical filters, and blood analysis sensors.[1] Photopolymers have also been explored for uses in drug delivery, tissue engineering and cell encapsulation systems.[15] Photopolymerization processes for these applications are being developed to be carried out in vivo or ex vivo. In vivo photopolymerization would provide the advantages of production and implantation with minimal invasive surgery.Ex vivo photopolymerization would allow for fabrication of complex matrices, and versatility of formulation. Although photopolymers show promise for a wide range of new biomedical applications, biocompatibility with photopolymeric materials must still be addressed and developed.
3D-imaging
Stereolithography, digital imaging, and 3D inkjet printing are just a few 3D imaging technologies that make use of photopolymers. 3D imaging usually proceeds with CAD-CAM software, which creates a 3D image to be translated into a 3D plastic object. The image is cut in slices, where each slice is reconstructed through radiation curing of the liquid polymer,converting the image into a solid object. Photopolymers used in 3D imaging processes must be designed to have a low volume shrinkage upon polymerization in order to avoid distortion of the solid object. Common monomers utilized for 3D imaging include multifunctional acrylates and methacrylates combined with a non-polymeric component in order to reduce volume shrinkage. A competing composite mixture of epoxide resins with cationic photoinitiators is becoming increasingly used since their volume shrinkage upon ring-opening polymerization is significantly below those of acrylates and methacrylates. Free-radical and cationic polymerizations composed of both epoxide and acrylate monomers have also been employed, gaining the high rate of polymerization from the acryilic monomer, and better mechanical properties from the epoxy matrix.[1]
Photoresists
Photoresists are coatings, or oligomers, that are deposited on a surface and are designed to change properties upon irradiation of light. These changes either polymerize the liquid oligomers into insoluble cross-linked network polymers or decompose the already solid polymers into liquid products. Polymers that form networks during photopolymerization are referred to as negative resist. Conversely, polymers that decompose during photopolymerization are referred to as positive resists. Both positive and negative resists have found many applications including the design and production of micro fabicated chips. The ability to pattern the resist using a focused light source has driven the field of photolithography.

Negative resists
As mentioned, negative resists are photopolymers that become insoluble upon exposure to radiation. They have found a variety of commercial applications. Especially in the area of designing and printing small chips for electronics. A characteristic found in most negative tone resists is the presence of multifunctional branches on the polymers used. Radiation of the polymers in the presence of an intiator results in the formation of chemically resistant network polymer. A common functional group used in negative resist is epoxy functional groups. An example of a widely used polymer of this class is SU-8. SU-8 was one of the first polymers used in this field, and found applications in wire board printing.[16] In the presence of a cationic photoinitiator photopolymer SU-8 forms networks with other polymers in solution. Basic scheme shown below.

SU-8 is an example of an intramolecular photopolymerization forming a matrix of cross-linked material. Negative resists can also be made using co-polymerization. In the event that you have two different monomers, or oligomers, in solution with multiple functionalities it is possible for the two to polymerize and form a less soluble polymer.
Positive resists
As mentioned, positive resist exposure to radiation changes the chemical structure such that it becomes a liquid or more soluble. These changes in chemical structure are often rooted in the cleavage of specific linkers in the polymer. Once irradiated the "decomposed" polymers can be washed away using a developer solvent leaving behind the polymer that was not exposed to light. This type of technology allows the production of very fine stencils for applications such as microelectronics.[17] In order to have these types of qualities, positive resist utilize polymers with labile linkers in their back bone that can be cleaved upon irradiation or using a photo-generated acid to hydrolyze bonds in the polymer. A polymer that decomposes upon irradiation to a liquid, or more soluble product is referred to as a positive tone resist. Common functional groups that can be hydrolyzed by photo-generated acid catalyst include polycarbonates and polyesters.[18]
Fine printing

Photopolymer can be used to generate printing plates, which are then pressed onto paper like metal type.[19] This is often used in modern fine printing to achieve the effect of embossing (or the more subtly three-dimensional effect of letterpress printing) from designs created on a computer without needing to engrave designs into metal or cast metal type. It is often used for business cards.[20][21]
References
- 1 2 3 4 5 6 7 Reichmanis, Elsa; Crivello, James (2014). "Photopolymer Materials and Processes for Advanced Technologies". Chem. Mater. 26: 533–548.
- ↑ Phillips, Roger (1984). "Photopolymerization". Journal of Photopolymerization. 25: 79–82. doi:10.1016/0047-2670(84)85016-9.
- ↑ Burton, Jeff. "A Primer on UV-Curable Inkjet Inks". Specialty Graphic Imaging Association.
- 1 2 3 4 5 6 Ravve, A. (2006). Light-Associated Reactions of Synthetic Polymers. Spring Street, New York, NY 10013, USA: Springer Science+Business Media, LLC. ISBN 0-387-31803-8.
- 1 2 3 4 5 6 Fouassier, Jean Pierre (2012). Photoinitiators for Polymer Synthesis: Scope, Reactivity and Efficiency. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA. ISBN 9783527648245.
- ↑ "Radiation Chemistry in EB-and UV-Light Cured Inks". Paint & Coatings Industry.
- 1 2 Fouassier, J.P.; Allonas, X.; Burget, D. (2003). "Photopolyermziation reactions under visible lights: principle, mechanisms and examples of applications". Progress in Organic Coatings. 47: 16–36. doi:10.1016/S0300-9440(03)00011-0.
- ↑ Cowie, J.M.G. (2008). Polymers: Chemistry and Physics of Modern Materials. CRC Press: Taylor and Francis Group. p. 76.
- 1 2 Crivello, J.; E. Reichmanis (2014). "Photopolymer Materials and Processes for Advanced Technologies". Chemistry of Materials. 26: 533–548. doi:10.1021/cm402262g.
- ↑ Zhdankin, Viktor (2013). Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds. John Wiley & Sons Ltd. p. 427.
- ↑ Fouassier, Jean (2012). Photoinitiators for Polymer Synthesis: Scope, Reactivity, and Efficiency. John Wiley & Sons Ltd. p. 293.
- ↑ Meier, K (1985). Proceedings of the RadCure Europe. Basle Technical Paper.
- ↑ TAKAHASHI, EIJI; FUMIO SANDA; TAKESHI ENDO (2002). "Novel pyridinium salts as cationic thermal and photoinitiators and their photosensitization properties". Journal of Polymer Science Part A: Polymer Chemistry. 40 (8): 1037. doi:10.1002/pola.10186.
- 1 2 3 Hoyle, Charles (1990). Radiation Curing of Polymeric Materials. Washington, DC: Am. Chem. Soc. pp. 1–15.
- ↑ Baroli, Biancamaria (2006). "Photopolymerization of biomaterials". J. Chem. Technol. Biotechnol. 81: 491–499. doi:10.1002/jctb.1468.
- ↑ "SU-8 Photosensitive Epoxy". Retrieved 2014. Check date values in:
|access-date=(help) - ↑ Allcock, Harry (2008). Introduction to Materials Chemistry. Wiley and Sons. pp. 248–258. ISBN 9780470293331.
- ↑ Thompson, Larry (1993). Polymers for Microelectronics. American Chemical Society.
- ↑ "What is a "faux-emboss"?". Dolce Press. Retrieved 24 September 2015.
- ↑ "Letterpress polymer plate service". Old City Press. Retrieved 24 September 2015.
- ↑ "What is Letterpress?". Baltimore Print Studios. Retrieved 24 September 2015.