Soft-tissue sarcoma
| Soft-tissue sarcoma | |
|---|---|
| Classification and external resources | |
| ICD-9-CM | 171 |
| DiseasesDB | 31472 |
| MeSH | D018204 |
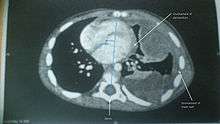
A soft-tissue sarcoma is a form of sarcoma that develops in connective tissue,[1] though the term is sometimes applied to elements of the soft tissue that are not currently considered connective tissue.
Risk factors
Most soft-tissue sarcomas are not associated with any known risk factors or identifiable etiology. There are some exceptions:
- Studies suggest that workers who are exposed to chlorophenols in wood preservatives and phenoxy herbicides may have an increased risk of developing soft-tissue sarcomas. An unusual percentage of patients with a rare blood vessel tumor, angiosarcoma of the liver, have been exposed to vinyl chloride in their work. This substance is used in the manufacture of certain plastics, notably PVC.
- In the early 1900s, when scientists were just discovering the potential uses of radiation to treat disease, little was known about safe dosage levels and precise methods of delivery. At that time, radiation was used to treat a variety of noncancerous medical problems, including enlargement of the tonsils, adenoids, and thymus gland. Later, researchers found that high doses of radiation caused soft-tissue sarcomas in some patients.[2] Because of this risk, radiation treatment for cancer is now planned to ensure that the maximum dosage of radiation is delivered to diseased tissue while surrounding healthy tissue is protected as much as possible.
- Kaposi's sarcoma, a rare cancer of the cells that line blood vessels in the skin and mucus membranes, is caused by Human herpesvirus 8. Kaposi's sarcoma often occurs in patients with AIDS (acquired immune deficiency syndrome). Kaposi's sarcoma, however, has different characteristics than typical soft-tissue sarcomas and is treated differently.
- In a very small fraction of cases, sarcoma may be related to a rare inherited genetic alteration of the p53 gene and is known as Li-Fraumeni syndrome. Certain other inherited diseases are associated with an increased risk of developing soft-tissue sarcomas. For example, people with neurofibromatosis type I (also called von Recklinghausen's disease, associated with alterations in the NF1 gene) are at an increased risk of developing soft-tissue sarcomas known as malignant peripheral nerve sheath tumors. Patients with inherited retinoblastoma have alterations in the RB1 gene, a tumor suppressor gene, and are likely to develop soft-tissue sarcomas as they mature into adulthood.
Epidemiology
Soft-tissue sarcomas are relatively uncommon cancers. They account for less than 1% of all new cancer cases each year. This may be because cells in soft tissue, in contrast to tissues that more commonly give rise to malignancies, are not continuously dividing cells.
In 2006, about 9,500 new cases were diagnosed in the United States.[3] Soft-tissue sarcomas are more commonly found in older patients (>50 years old) although in children and adolescents under age 20, certain histologies are common (rhabdomyosarcoma, synovial sarcoma).
Around 3,300 people were diagnosed with soft tissue sarcoma in the UK 2011.[4]
Symptoms
In their early stages, soft-tissue sarcomas usually do not cause symptoms. Because soft tissue is relatively elastic, tumors can grow rather large, pushing aside normal tissue, before they are felt or cause any problems. The first noticeable symptom is usually a painless lump or swelling. As the tumor grows, it may cause other symptoms, such as pain or soreness, as it presses against nearby nerves and muscles. If in the abdomen it can cause abdominal pains commonly mistaken for menstrual cramps, indigestion, or cause constipation.
Diagnosis
The only reliable way to determine whether a soft-tissue tumour is benign or malignant is through a biopsy. There are two methods for acquisition of tumour tissue for cytopathological analysis;
- Needle Aspiration, via biopsy needle
- surgically, via an incision made into the tumour.
A pathologist examines the tissue under a microscope. If cancer is present, the pathologist can usually determine the type of cancer and its grade. Here, 'grade' refers to a scale used to represent concisely the predicted growth rate of the tumour and its tendency to spread, and this is determined by the degree to which the cancer cells appear abnormal when examined under a microscope. Low-grade sarcomas, although cancerous, are defined as those that are less likely to metastasise. High-grade sarcomas are defined as those more likely to spread to other parts of the body.
Soft tissue sarcomas commonly originate in the upper body, in the shoulder or upper chest. Some symptoms are uneven posture, pain in the trapezius muscle and cervical inflexibility [difficulty in turning the head].
The most common site to which soft tissue sarcoma spreads is the lungs.
Treatment
In general, treatment for soft-tissue sarcomas depends on the stage of the cancer. The stage of the sarcoma is based on the size and grade of the tumor, and whether the cancer has spread to the lymph nodes or other parts of the body (metastasized). Treatment options for soft-tissue sarcomas include surgery, radiation therapy, and chemotherapy.
- Surgery is the most common treatment for soft-tissue sarcomas. If possible, the doctor will remove the cancer and a safe margin of the healthy tissue around it. It is important to obtain a margin free of tumor to decrease the likelihood of local recurrence and give the best chance for eradication of the tumor. Depending on the size and location of the sarcoma, it may, rarely, be necessary to remove all or part of an arm or leg.
- Radiation therapy may be used either before surgery to shrink tumors or after surgery to kill any cancer cells that may have been left behind. In some cases, it can be used to treat tumours that cannot be surgically removed. In multiple studies, radiation therapy has been found to improve the rate of local control, but has not had any influence on overall survival.
- Chemotherapy may be used with radiation therapy either before or after surgery to try to shrink the tumor or kill any remaining cancer cells. The use of chemotherapy to prevent the spread of soft-tissue sarcomas has not been proven to be effective. If the cancer has spread to other areas of the body, chemotherapy may be used to shrink tumors and reduce the pain and discomfort they cause, but is unlikely to eradicate the disease.
A combination of Taxotere and Gemzar could be an effective chemotherapy regimen in patients with advanced soft-tissue sarcoma.[5]
Tables
| Tissue of Origin | Type of Cancer | Usual Location in the Body |
|---|---|---|
| Fibrous tissue | Fibrosarcoma | Arms, legs, trunk |
| Malignant fibrous hystiocytoma | Legs | |
| Dermatofibrosarcoma | Trunk | |
| Fat | Liposarcoma | Arms, legs, trunk |
| Muscle |
Rhabdomyosarcoma Leiomyosarcoma |
Arms, legs Uterus, digestive tract |
| Blood vessels | Hemangiosarcoma | Arms, legs, trunk |
| Kaposi's sarcoma | Legs, trunk | |
| Lymph vessels | Lymphangiosarcoma | Arms |
| Synovial tissue (linings of joint cavities, tendon sheaths) |
Synovial sarcoma | Legs |
| Peripheral nerves | Malignant peripheral nerve sheath tumour/Neurofibrosarcoma | Arms, legs, trunk |
| Cartilage and bone-forming tissue | Extraskeletal chondrosarcoma | Legs |
| Extraskeletal osteosarcoma | Legs, trunk (not involving the bone) |
| Tissue of Origin | Type of Cancer | Usual Location in the Body | Most common ages |
|---|---|---|---|
| Muscle | |||
| |
Rhabdomyosarcoma | ||
| |
Head and neck, genitourinary tract | Infant–4 | |
| |
Arms, legs, head, and neck | Infant–19 | |
| |
Leiomyosarcoma | Trunk | 15–19 |
| Fibrous tissue | Fibrosarcoma | Arms and legs | 15–19 |
| Malignant fibrous histiocytoma |
Legs | 15–19 | |
| Dermatofibrosarcoma | Trunk | 15–19 | |
| Fat | Liposarcoma | Arms and Legs | 15–19 |
| Blood vessels | Infantile hemangio- |
Arms, legs, trunk, head, and neck | Infant–4 |
| Synovial tissue (linings of joint cavities, tendon sheaths) |
Synovial sarcoma | Legs, arms, and trunk | 15–19 |
| Peripheral nerves | Malignant peripheral nerve sheath tumors (also called neurofibrosarcomas, malignant schwannomas, and neurogenic sarcomas) | Arms, legs, and trunk | 15–19 |
| Muscular nerves | Alveolar soft part sarcoma | Arms and legs | Infant–19 |
| Cartilage and bone-forming tissue | Extraskeletal myxoid chondrosarcoma | Legs | 10–14 |
| Extraskeletal mesenchymal | Legs | 10–14 |
An earlier version of this article was taken from the US National Cancer Center's Cancer Information Service.
Notable patients
- Actor Robert Urich died from synovial sarcoma.
- Actress Michelle Thomas died from desmoplastic small-round-cell tumor, a rare abdominal soft-tissue sarcoma.
- It Is Written evangelist Henry Feyerabend died from sarcoma in his leg.
- Video game concept artist Adam Adamowicz died from complications of a rare muscle sarcoma on Feb. 9, 2012. He was 43.
- Professional wrestler, Jake Roberts revealed he is afflicted with muscle cancer.
- Professional wrestler, Zack Ryder revealed he suffered from Synovial Sarcoma as a teenager.[6]
References
- ↑ "soft tissue sarcoma" at Dorland's Medical Dictionary
- ↑ Dino Samartzis, University of Hong Kong, South China Morning Post, 1 June 2013
- ↑ Ries LAG, Harkins D, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2003. Bethesda , MD: National Cancer Institute, 2006.
- ↑ "Soft tissue sarcoma statistics". Cancer Research UK. Retrieved 28 October 2014.
- ↑ http://professional.cancerconsultants.com/oncology_sarcoma_cancer_news.aspx?id=36670
- ↑ http://www.wwe.com/inside/the-scars-of-the-superstars/page-13
External links
- Factsheet from the National Cancer Institute
- Information from Memorial Sloan Kettering
- The Sarcoma Learning Center
- www.soft-tissue.com - a database of review questions on soft tissue sarcomas
- Clinically reviewed soft-tissue sarcoma information for patients, from Cancer Research UK.
- OrthoTumours a case-based educational resource