VSEPR theory
Valence shell electron pair repulsion (VSEPR) theory is a model used in chemistry to predict the geometry of individual molecules from the number of electron pairs surrounding their central atoms.[1] It is also named the Gillespie-Nyholm theory after its two main developers. The acronym "VSEPR" is pronounced either "ves-per"[2]:410 or "vuh-seh-per"[3] by some chemists.
The premise of VSEPR is that the valence electron pairs surrounding an atom tend to repel each other, and will therefore adopt an arrangement that minimizes this repulsion, thus determining the molecule's geometry. Gillespie has emphasized that the electron-electron repulsion due to the Pauli exclusion principle is more important in determining molecular geometry than the electrostatic repulsion.[4]
VSEPR theory is based on observable electron density rather than mathematical wave functions and hence unrelated to orbital hybridisation,[5] although both address molecular shape. While it is mainly qualitative, VSEPR has a quantitative basis in quantum chemical topology (QCT) methods such as the electron localization function and the quantum theory of atoms in molecules (QTAIM).[4]
History
The idea of a correlation between molecular geometry and number of valence electrons (both shared and unshared) was originally proposed in 1939 by Ryutaro Tsuchida in Japan,[6] and was independently presented in a Bakerian Lecture in 1940 by Nevil Sidgwick and Herbert Powell of the University of Oxford.[7] In 1957, Ronald Gillespie and Ronald Sydney Nyholm of University College London refined this concept into a more detailed theory, capable of choosing between various alternative geometries.[8][9]
Overview
VSEPR theory is used to predict the arrangement of electron pairs around non-hydrogen atoms in molecules, especially simple and symmetric molecules, where these key, central atoms participate in bonding to two or more other atoms; the geometry of these key atoms and their non-bonding electron pairs in turn determine the geometry of the larger whole.
The number of electron pairs in the valence shell of a central atom is determined after drawing the Lewis structure of the molecule, and expanding it to show all bonding groups and lone pairs of electrons.[2]:410–417 In VSEPR theory, a double bond or triple bond are treated as a single bonding group.[2] The sum of the number of atoms bonded to a central atom and the number of lone pairs formed by its nonbonding valence electrons is known as the central atom's steric number.
The electron pairs (or groups if double, triple... bonds are present) are assumed to lie on the surface of a sphere centered on the central atom and tend to occupy positions that minimize their mutual repulsions by maximizing the distance between them.[2]:410–417[10] The number of electron pairs (or groups), therefore, determines the overall geometry that they will adopt. For example, when there are two electron pairs surrounding the central atom, their mutual repulsion is minimal when they lie at opposite poles of the sphere. Therefore, the central atom is predicted to adopt a linear geometry. If there are 3 electron pairs surrounding the central atom, their repulsion is minimized by placing them at the vertices of an equilateral triangle centered on the atom. Therefore, the predicted geometry is trigonal. Likewise, for 4 electron pairs, the optimal arrangement is tetrahedral.[2]:410–417
Degree of repulsion
The overall geometry is further refined by distinguishing between bonding and nonbonding electron pairs. The bonding electron pair shared in a sigma bond with an adjacent atom lies further from the central atom than a nonbonding (lone) pair of that atom, which is held close to its positively charged nucleus. VSEPR theory therefore views repulsion by the lone pair to be greater than the repulsion by a bonding pair. As such, when a molecule has 2 interactions with different degrees of repulsion, VSEPR theory predicts the structure where lone pairs occupy positions that allow them to experience less repulsion. Lone pair–lone pair (lp–lp) repulsions are considered stronger than lone pair–bonding pair (lp–bp) repulsions, which in turn are considered stronger than bonding pair–bonding pair (bp–bp) repulsions, distinctions that then guide decisions about overall geometry when 2 or more non-equivalent positions are possible.[2]:410–417 For instance, when 5 valence electron pairs surround a central atom, they adopt a trigonal bipyramidal molecular geometry with two collinear axial positions and three equatorial positions. An electron pair in an axial position has three close equatorial neighbors only 90° away and a fourth much farther at 180°, while an equatorial electron pair has only two adjacent pairs at 90° and two at 120°. The repulsion from the close neighbors at 90° is more important, so that the axial positions experience more repulsion than the equatorial positions; hence, when there are lone pairs, they tend to occupy equatorial positions as shown in the diagrams of the next section for steric number five.[10]
The difference between lone pairs and bonding pairs may also be used to rationalize deviations from idealized geometries. For example, the H2O molecule has four electron pairs in its valence shell: two lone pairs and two bond pairs. The four electron pairs are spread so as to point roughly towards the apices of a tetrahedron. However, the bond angle between the two O–H bonds is only 104.5°, rather than the 109.5° of a regular tetrahedron, because the two lone pairs (whose density or probability envelopes lie closer to the oxygen nucleus) exert a greater mutual repulsion than the two bond pairs.[2]:410–417[10]
An advanced-level explanation replaces the above distinction with two rules:
- Bent's rule: An electron pair of a more electropositive ligand constitutes greater repulsion. This explains why the Cl in PClF4 prefers the equatorial position and why the bond angle in oxygen difluoride (103.8°) is smaller than that of water (104.5°). Lone pairs are then considered to be a special case of this rule, held by a "ghost ligand" in the limit of electropositivity.
- A higher bond order constitutes greater repulsion. This explains why in phosgene, the oxygen–chlorine bond angle (124.1°) is larger than the chlorine–chlorine bond angle (111.8°) even though chlorine is more electropositive than oxygen. In the carbonate ion, all three bond angles are equivalent due to resonance.
AXE method
The "AXE method" of electron counting is commonly used when applying the VSEPR theory. The A represents the central atom and always has an implied subscript one. The X represents each of ligands (atoms bonded to A). The E represents the number of lone electron pairs surrounding the central atom.[2]:410–417 The sum of X and E is known as the steric number.
Based on the steric number and distribution of Xs and Es, VSEPR theory makes the predictions in the following tables. Note that the geometries are named according to the atomic positions only and not the electron arrangement. For example, the description of AX2E1 as a bent molecule means that the three atoms AX2 are not in one straight line, although the lone pair helps to determine the geometry.
| Steric number |
Molecular geometry[11] 0 lone pairs |
Molecular geometry[2]:413–414 1 lone pair |
Molecular geometry[2]:413–414 2 lone pairs |
Molecular geometry[2]:413–414 3 lone pairs |
|---|---|---|---|---|
| 2 | | |||
| 3 |  | 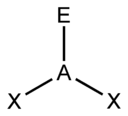 | ||
| 4 | 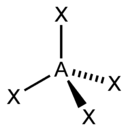 |  | 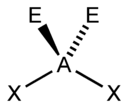 | |
| 5 |  |  | 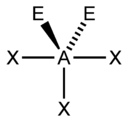 |  3) |
| 6 |  | 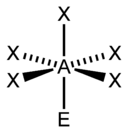 |  | |
| 7 | 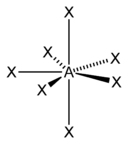 | 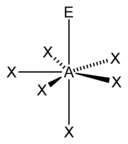 5)[12] |  5)[13]:498 | |
| 8 | (TaF3− 8)[10] | | ||
| 9 | Tricapped trigonal prismatic (ReH2− 9)[13]:254 or Capped square antiprismatic | |||
| 10 | Bicapped square antiprismatic or Bicapped dodecadeltahedral[14]:1165,1721 | |||
| 11 | Octadecahedral[14]:1165,1721 | |||
| 12 | Icosahedral[14]:1165,1721 | |||
| 14 | Bicapped hexagonal antiprismatic[14]:1165,1721 |



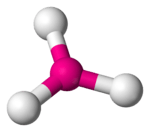



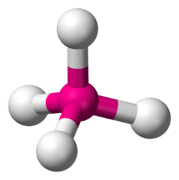


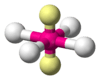


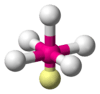


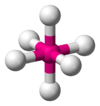

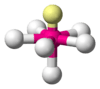
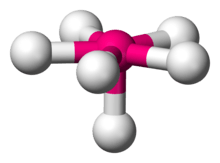





When the substituent (X) atoms are not all the same, the geometry is still approximately valid, but the bond angles may be slightly different from the ones where all the outside atoms are the same. For example, the double-bond carbons in alkenes like C2H4 are AX3E0, but the bond angles are not all exactly 120°. Likewise, SOCl2 is AX3E1, but because the X substituents are not identical, the X–A–X angles are not all equal.
As a tool in predicting the geometry adopted with a given number of electron pairs, an often used physical demonstration of the principle of minimal electron pair repulsion utilizes inflated balloons. Through handling, balloons acquire a slight surface electrostatic charge that results in the adoption of roughly the same geometries when they are tied together at their stems as the corresponding number of electron pairs. For example, five balloons tied together adopt the trigonal bipyramidal geometry, just as do the five bonding pairs of a PCl5 molecule (AX5) or the two bonding and three non-bonding pairs of a XeF2 molecule (AX2E3). The molecular geometry of the former is also trigonal bipyramidal, whereas that of the latter is linear.
Examples
The methane molecule (CH4) is tetrahedral because there are four pairs of electrons. The four hydrogen atoms are positioned at the vertices of a tetrahedron, and the bond angle is cos−1(− 1⁄3) ≈ 109° 28′.[15][16] This is referred to as an AX4 type of molecule. As mentioned above, A represents the central atom and X represents an outer atom.[2]:410–417
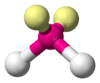
The ammonia molecule (NH3) has three pairs of electrons involved in bonding, but there is a lone pair of electrons on the nitrogen atom.[2]:392–393 It is not bonded with another atom; however, it influences the overall shape through repulsions. As in methane above, there are four regions of electron density. Therefore, the overall orientation of the regions of electron density is tetrahedral. On the other hand, there are only three outer atoms. This is referred to as an AX3E type molecule because the lone pair is represented by an E.[2]:410–417 By definition, the molecular shape or geometry describes the geometric arrangement of the atomic nuclei only, which is trigonal-pyramidal for NH3.[2]:410–417
Steric numbers of 7 or greater are possible, but are less common. The steric number of 7 occurs in iodine heptafluoride (IF7); the base geometry for a steric number of 7 is pentagonal bipyramidal.[10] The most common geometry for a steric number of 8 is a square antiprismatic geometry.[14]:1165 Examples of this include the octacyanomolybdate (Mo(CN)4−
8) and octafluorozirconate (ZrF4−
8) anions.[14]:1165
The nonahydridorhenate ion (ReH2−
9) in potassium nonahydridorhenate is a rare example of a compound with a steric number of 9, which has a tricapped trigonal prismatic geometry.[13]:254[14] Another example is the octafluoroxenate ion (XeF2−
8) in nitrosonium octafluoroxenate(VI),[13]:498[17][18] although in this case one of the electron pairs is a lone pair, and therefore the molecule actually has a distorted square antiprismatic geometry.
Possible geometries for steric numbers of 10, 11, 12, or 14 are bicapped square antiprismatic (or bicapped dodecadeltahedral), octadecahedral, icosahedral, and bicapped hexagonal antiprismatic, respectively. No compounds with steric numbers this high involving monodentate ligands exist, and those involving multidentate ligands can often be analysed more simply as complexes with lower steric numbers when some multidentate ligands are treated as a unit.[14]:1165,1721
Exceptions
There are groups of compounds where VSEPR fails to predict the correct geometry.
Some AX2E0 molecules
The gas phase structures of the triatomic halides of the heavier members of group 2, (i.e., calcium, strontium and barium halides, MX2), are not linear as predicted but are bent, (approximate X–M–X angles: CaF2, 145°; SrF2, 120°; BaF2, 108°; SrCl2, 130°; BaCl2, 115°; BaBr2, 115°; BaI2, 105°).[19] It has been proposed by Gillespie that this is caused by interaction of the ligands with the electron core of the metal atom, polarising it so that the inner shell is not spherically symmetric, thus influencing the molecular geometry.[20][21] Ab initio calculations have been cited to propose that contributions from d orbitals in the shell below the valence shell are responsible.[22] Disilynes are also bent, despite having no lone pairs.[23]
Some AX2E2 molecules
One example of the AX2E2 geometry is molecular lithium oxide, Li2O, a linear rather than bent structure, which is ascribed to its bonds being essentially ionic and the strong lithium-lithium repulsion that results.[24] Another example is O(SiH3)2 with an Si–O–Si angle of 144.1°, which compares to the angles in Cl2O (110.9°), (CH3)2O (111.7°), and N(CH3)3 (110.9°).[20] Gillespie and Robinson rationalize the Si–O–Si bond angle based on the observed ability of a ligand's lone pair to most greatly repel other electron pairs when the ligand electronegativity is greater than or equal to that of the central atom.[20] In O(SiH3)2, the central atom is more electronegative, and the lone pairs are less localized and more weakly repulsive. The larger Si–O–Si bond angle results from this and strong ligand-ligand repulsion by the relatively large -SiH3 ligand.[20]
Some AX6E1 and AX8E1 molecules

Some AX6E1 molecules, e.g. xenon hexafluoride (XeF6) and the Te(IV) and Bi(III) anions, TeCl2−
6, TeBr2−
6, BiCl3−
6, BiBr3−
6 and BiI3−
6, are octahedra, rather than pentagonal pyramids, and the lone pair does not affect the geometry to the degree predicted by VSEPR.[25] One rationalization is that steric crowding of the ligands allows little or no room for the non-bonding lone pair;[20] another rationalization is the inert pair effect.[13]:214
Transition metal molecules

Many transition metal compounds have unusual geometries, which can be ascribed to ligand bonding interaction with the d subshell and to absence of valence shell lone pairs.[26] Gillespie suggested that this interaction can be weak or strong. Weak interaction is dealt with by the Kepert model, while strong interaction produces bonding pairs that also occupy the respective antipodal points of the sphere.[4] This is similar to predictions based on sd hybrid orbitals[27][28] using the VALBOND theory. The repulsion of these bidirectional bonding pairs leads to a different prediction of shapes.
| Molecule type | Shape | Geometry | Examples |
|---|---|---|---|
| AX2 | Bent |  |
VO+ 2 |
| AX3 | Trigonal pyramidal | 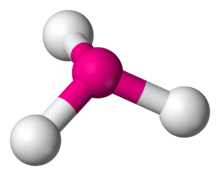 |
CrO3 |
| AX4 | Tetrahedral | 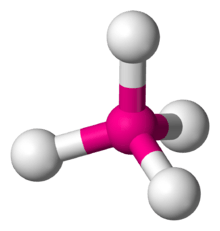 |
TiCl4[13]:598–599 |
| AX5 | Square pyramidal |  |
Ta(CH3)5[29] |
| AX6 | Trigonal prismatic | 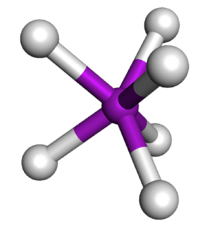 |
W(CH3)6[30] |
The square planar shape associated with a d8 electronic configuration is an exception to the Kepert model. This can be rationalized by considering the increased crystal field stabilization energy as compared to a tetrahedral geometry.
Odd-electron molecules
The VSEPR theory can be extended to molecules with an odd number of electrons by treating the unpaired electron as a "half electron pair" — for example, Gillespie and Nyholm[8]:364–365 suggested that the decrease in the bond angle in the series NO+
2 (180°), NO2 (134°), NO−
2 (115°) indicates that a given set of bonding electron pairs exert a weaker repulsion on a single non-bonding electron than on a pair of non-bonding electrons. In effect, they considered nitrogen dioxide as an AX2E0.5 molecule with a geometry intermediate between NO+
2 and NO−
2. Similarly chlorine dioxide (ClO2) is AX2E1.5 with a geometry intermediate between ClO+
2 and ClO−
2.
Finally the methyl radical (CH3) is predicted to be trigonal pyramidal like the methyl anion (CH−
3), but with a larger bond angle as in the trigonal planar methyl cation (CH+
3). However, in this case the VSEPR prediction is not quite true, as CH3 is actually planar, although its distortion to a pyramidal geometry requires very little energy.[31]
See also
- Bent's rule (effect of ligand electronegativity)
- Linear combination of atomic orbitals
- Molecular geometry
- Molecular modelling
- Software for molecular modeling
- Thomson problem
- Valency interaction formula
References
- 1 2 3 4 5 6 7 Jolly, W. L. (1984). Modern Inorganic Chemistry. McGraw-Hill. p. 77–90. ISBN 0-07-032760-2.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 Petrucci, R. H.; W. S., Harwood; F. G., Herring (2002). General Chemistry: Principles and Modern Applications (8th ed.). Prentice-Hall. ISBN 0-13-014329-4.
- ↑ Stoker, H. Stephen (2009). General, Organic, and Biological Chemistry. Cengage Learning. p. 119. ISBN 978-0-547-15281-3.
- 1 2 3 Gillespie, R. J. (2008). "Fifty years of the VSEPR model". Coord. Chem. Rev. 252: 1315–1327. doi:10.1016/j.ccr.2007.07.007.
- ↑ Gillespie, R. J. (2004), "Teaching molecular geometry with the VSEPR model", J. Chem. Ed., 81 (3): 298–304, Bibcode:2004JChEd..81..298G, doi:10.1021/ed081p298
- ↑ Tsuchida, Ryutarō (1939). 新簡易原子價論 [New simple valency theory]. J. Chem. Soc. Jpn. (in Japanese). 60 (3): 245–256. doi:10.1246/nikkashi1921.60.245.
- ↑ Sidgwick, N. V.; Powell, H. M. (1940). "Bakerian Lecture. Stereochemical Types and Valency Groups". Proc. Roy. Soc. A. 176: 153–180. doi:10.1098/rspa.1940.0084.
- 1 2 Gillespie, R. J.; Nyholm, R. S. (1957). "Inorganic stereochemistry". Quart. Rev. Chem. Soc. 11: 339. doi:10.1039/QR9571100339.
- ↑ Gillespie, R. J. (1970). "The electron-pair repulsion model for molecular geometry". J. Chem. Educ. 47 (1): 18. doi:10.1021/ed047p18.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Miessler, G. L.; Tarr, D. A. (1999). Inorganic Chemistry (2nd ed.). Prentice-Hall. p. 54–62. ISBN 0-13-841891-8.
- ↑ Petrucci, R. H.; W. S., Harwood; F. G., Herring (2002). General Chemistry: Principles and Modern Applications (8th ed.). Prentice-Hall. p. 413–414 (Table 11.1). ISBN 0-13-014329-4.
- 1 2 3 Baran, E. (2000). "Mean amplitudes of vibration of the pentagonal pyramidal XeOF−
5 and IOF2−
5 anions". J. Fluorine Chem. 101: 61–63. doi:10.1016/S0022-1139(99)00194-3. - 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Housecroft, C. E.; Sharpe, A. G. (2005). Inorganic Chemistry (2nd ed.). Pearson. ISBN 978-0-130-39913-7.
- 1 2 3 4 5 6 7 8 Wiberg, E.; Holleman, A. F. (2001). Inorganic Chemistry. Academic Press. ISBN 0-12-352651-5.
- ↑ Brittin, W. E. (1945). "Valence Angle of the Tetrahedral Carbon Atom". J. Chem. Educ. 22 (3): 145. doi:10.1021/ed022p145.
- ↑ "Angle Between 2 Legs of a Tetrahedron" – Maze5.net
- ↑ Peterson, W.; Holloway, H.; Coyle, A.; Williams, M. (Sep 1971). "Antiprismatic Coordination about Xenon: the Structure of Nitrosonium Octafluoroxenate(VI)". Science. 173 (4003): 1238–1239. Bibcode:1971Sci...173.1238P. doi:10.1126/science.173.4003.1238. ISSN 0036-8075. PMID 17775218.
- ↑ Hanson, Robert M. (1995). Molecular origami: precision scale models from paper. University Science Books. ISBN 0-935702-30-X.
- ↑ Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 0-08-037941-9.
- 1 2 3 4 5 Gillespie, R. J.; Robinson, E. A. (2005). "Models of molecular geometry". Chem. Soc. Rev. 34: 396–407. doi:10.1039/b405359c.
- ↑ Bytheway, I.; Gillespie, R. J.; Tang, T. H.; Bader, R.F (1995). "Core Distortions and Geometries of the Difluorides and Dihydrides of Ca, Sr, and Ba". Inorg. Chem. 34 (9): 2407–2414. doi:10.1021/ic00113a023.
- ↑ Seijo, Luis; Barandiarán, Zoila; Huzinaga, Sigeru (1991). "Ab initio model potential study of the equilibrium geometry of alkaline earth dihalides: MX2 (M=Mg, Ca, Sr, Ba; X=F, Cl, Br, I)". J. Chem. Phys. 94 (5): 3762. doi:10.1063/1.459748.
- ↑ Sekiguchi, Akira; Kinjō, Rei; Ichinohe, Masaaki (September 2004). "A Stable Compound Containing a Silicon–Silicon Triple Bond" (PDF). Science. 305 (5691): 1755–1757. Bibcode:2004Sci...305.1755S. doi:10.1126/science.1102209. PMID 15375262.
- ↑ Bellert, D.; Breckenridge, W. H. (2001). "A spectroscopic determination of the bond length of the LiOLi molecule: Strong ionic bonding". J. Chem. Phys. 114: 2871. doi:10.1063/1.1349424.
- ↑ Wells, A. F. (1984). Structural Inorganic Chemistry (5th ed.). Oxford Science Publications. ISBN 0-19-855370-6.
- ↑ Kaupp, Martin (2001). ""Non-VSEPR" Structures and Bonding in d0 Systems". Angew. Chem. Int. Ed. Engl. 40 (1): 3534–3565. doi:10.1002/1521-3773(20011001)40:19<3534::AID-ANIE3534>3.0.CO;2-#.
- ↑ Landis, C. R.; Cleveland, T.; Firman, T. K. (1995). "Making sense of the shapes of simple metal hydrides". J. Am. Chem. Soc. 117: 1859–1860. doi:10.1021/ja00111a036.
- ↑ Landis, C. R.; Cleveland, T.; Firman, T. K. (1996). "Structure of W(CH3)6". Science. 272: 179–183. doi:10.1126/science.272.5259.179f.
- ↑ King, R. Bruce (2000). "Atomic orbitals, symmetry, and coordination polyhedra". Coord. Chem. Rev. 197: 141–168. doi:10.1016/s0010-8545(99)00226-x.
- ↑ Haalan, A.; Hammel, A.; Rydpal, K.; Volden, H. V. (1990). "The coordination geometry of gaseous hexamethyltungsten is not octahedral". J. Am. Chem. Soc. 112 (11): 4547–4549. doi:10.1021/ja00167a065.
- ↑ Anslyn, E. V.; Dougherty, D. A. (2006). Modern Physical Organic Chemistry. University Science Books. p. 57. ISBN 978-1891389313.
Further reading
- Lagowski, J. J., ed. (2004). Chemistry: Foundations and Applications. 3. New York: Macmillan. p. 99–104. ISBN 0-02-865721-7.
External links
| The Wikibook A-level Chemistry/OCR (Salters) has a page on the topic of: Molecular geometry and lone pairs |
- 3D Chem – Chemistry, structures, and 3D Molecules
- IUMSC – Indiana University Molecular Structure Center