CO-methylating acetyl-CoA synthase
| CO-methylating acetyl-CoA synthase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 Monomeric Acetyl-CoA Synthase | |||||||||
| Identifiers | |||||||||
| EC number | 2.3.1.169 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / EGO | ||||||||
| |||||||||
Acetyl-CoA Synthase (ACS), not to be confused with Acetyl-CoA Synthetase or Acetate-CoA Ligase (ADP forming), is a Nickel containing enzyme involved in the metabolic processes of cells. Together with Carbon monoxide dehydrogenase (CODH), it forms the bifunctional enzyme Acetyl-CoA Synthase/Carbon Monoxide Dehydrogenase (ACS/CODH) found in anaerobic organisms such as archaea and eubacteria.[1] The ACS/CODH enzyme works primarily through the Wood–Ljungdahl pathway which converts Carbon dioxide to Acetyl-CoA. Another name for this enzyme is CO-methylating acetyl-CoA synthase.
Chemistry
In nature, there are six different pathways where CO2 is fixed. Of these, the Wood–Ljungdahl pathway is the predominant sink in anaerobic conditions. Acetyl-CoA Synthase (ACS) and carbon monoxide dehydrogenase (CODH) are integral enzymes in this one pathway and can perform diverse reactions in the carbon cycle as a result. Because of this, the exact activity of these molecules has come under intense scrutiny over the past decade.[2]
Wood-Ljungdahl pathway
The Wood–Ljungdahl pathway consists of two different reactions that break down carbon dioxide. The first pathway involves CODH converting carbon dioxide into carbon monoxide through a two-electron transfer, and the second reaction involves ACS synthesizing acetyl-CoA using the carbon monoxide from CODH together with coenzyme-A (CoA) and a methyl group from a corrinoid-iron sulfur protein, CFeSP.[3] The two main overall reactions are as follows:
-
(1)

-
(2)

The Acetyl-CoA produced can be used in a variety of ways depending on the needs of the organism. For example, acetate-forming bacteria use acetyl-CoA for their autotrophic growth processes, and methanogenic archae such as Methanocarcina barkeri convert the acetyl-CoA into acetate and use it as an alternative source of carbon instead of CO2.[4]

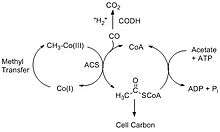
Since the above two reactions are reversible, it opens up a diverse range of reactions in the carbon cycle. In addition to acetyl-CoA production, the reverse can occur with ACS producing acetate, CO and returning the methyl piece back to the corrinoid protein. Acetogenic bacteria use this method to generate acetate and acetic acid. Along with the process of methanogenesis, organisms can subsequently convert the acetate to methane. Furthermore, the Wood-Ljungdahl pathway allows for the anaerobic oxidation of acetate where ATP is used to convert acetate into acetyl-CoA, which is then broken down by ACS to produce carbon dioxide that is released into the atmosphere.[5]
Other reactions
It has been discovered that the CODH/ACS enzyme in the bacteria M. theroaceticum can make dinitrogen (N2) from nitrous oxide in the presence of an electron-donating species. It can also catalyze the reduction of the pollutant, 2,4,6-trinitrotoluene (TNT) and catalyze the oxidation of n-butyl isocyanide.[2]
Structure
History
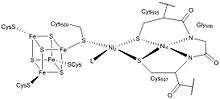
The first, and one of the most comprehensive, crystal structures of ACS/CODH from the bacteria M. thermoacetica was presented in 2002 by Drennan and colleagues.[6] In this paper they constructed a heterotetramer, with the active site "A-cluster" residing in the ACS subunit and the active site "C-cluster" in CODH subunit. Furthermore, they resolved the structure of the A-cluster active site and found an [Fe4S4]-X-Cu-X-Ni centre which is highly unusual in biology. This structural representation consisted of a [Fe4S4] unit bridged to a binuclear centre, where Ni(II) resided in the distal position (denoted as Nid) in a square-planar conformation and a Cu(I) ion resided in the proximal position in a distorted tetrahedral position with ligands of unknown identity.[6]
The debate towards the absolute structure and identity of the metals in the A-cluster active site of ACS continued, with a competing model presented. The authors suggested two different forms of the ACS enzyme, an "Open" form and a "Closed" form, with different metals occupying the proximal metal site (denoted as Mp) for each form. The general scheme of the enzyme followed closely with the first study's findings, but this new structure proposed a Nickel ion in the "open" form and a Zinc ion in the "closed" form.[3]
A later review article attempted to reconcile the different observations of Mp and stated that this proximal position in the active site of ACS was prone to substitution and could contain any one of Cu, Zn and Ni. The three forms of this A-cluster most likely hold a small amount of Ni and a relatively larger amount of Cu.[7]
Present (2014 onwards)
It is now generally accepted that the ACS active site (A-cluster) is a Ni-Ni metal centre with both nickels having a +2 oxidation state. The [Fe4S4] cluster is bridged to the closer nickel, Np which is connected via a thiolate bridge to the farther nickel, Nid. Nid is coordinated to two cysteine molecules and two backbone amide compounds, and is in a square-planar coordination. The space next to the metal can accommodate substrates and products. Nip is in a T-shaped environment bound to three sulfur atoms, with an unknown ligand possibly creating a distorted tetrahedral environment. This ligand has been hypothesized to be a water molecule or an acetyl group in the surrounding area in the cell. Although the proximal nickel is labile and can be replaced with a Cu of Zn centre, experimental evidence suggests that activity of ACS is limited to the presence of nickel only. In addition, some studies have shown that copper can even inhibit the enzyme under certain conditions.[8]
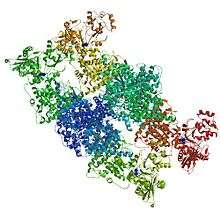
The overall structure of the CODH/ACS enzyme consists of the CODH enzyme as a dimer at the centre with two ACS subunits on each side. The CODH core is made up of two Ni-Fe-S clusters (C-cluster), two [Fe4S4] clusters (B-cluster) and one [Fe4S4] D-cluster. The D-cluster bridges the two subunits with one C and one B cluster in each monomer, allowing rapid electron transfer. The A-cluster of ACS is in constant communication with the C-cluster in CODH. This active site is also responsible for the C-C and C-S bond formations in the product acetyl-CoA (and its reverse reaction).[7]
The ACS enzyme contains three main subunits. The first is the active site itself with the NiFeS centre. The second is the portion that directly interacts with CODH in the Wood-Ljungdahl pathway. This part is made up of α-helices that go into a Rossman fold. It also appears to interact with a ferredoxin compound which may activate the subunit during the CO transferring process from CODH to ACS. The final domain binds CoA and consists of six arginine residues with a tryptophan molecule.[2][9]
Experiments between the C-cluster of CODH and the A-cluster of ACS reveal a long, hydrophobic channel connecting the two domains to allow for the transfer of carbon monoxide from CODH to ACS. This channel is most likely to protect the carbon monoxide molecules from the outside environment of the enzyme and to increase efficiency of acetyl-CoA production.[10]
Conformational changes
Studies in literature have been able to isolate the CODH/ACS enzyme in an "open" and "closed" configuration. This has led to the hypothesis that it undergoes four conformational changes depending on its activity. With the "open" position, the active site rotates itself to interact with the CFeSP protein in the methyl transfer step of the Wood-Ljungdahl pathway. The "closed" position opens up the channel between CODH and ACS to allow for the transfer of CO. These two configurations are opposite one another in that access to CO blocks off interaction with CFeSP, and when methylation occurs, the active site is buried and does not allow CO transfer. A second "closed" position is needed to block off water from the reaction. Finally, the A-cluster must be rotated once more to allow for the binding of CoA and release of the product. The exact trigger of these structural changes and the mechanistic details have yet to be resolved.[2][5][8]
Activity
Mechanism

Two competing mechanisms have been proposed for the formation of acetyl-CoA, the "Paramagnetic mechanism" and the "Diamagnetic mechanism".[2] Both are similar in terms of the binding of substrates and the general steps, but differ in the oxidation state of the metal centre. Nip is believed to be the substrate binding centre which undergoes redox. The farther nickel centre and the [Fe4S4] cluster are not thought to be involved in the process.[10]
In the paramagnetic mechanism, some type of complex (ferrodoxin, for example) activates the Nip atom, reducing it from Ni2+ to Ni1+. The nickel then binds to either carbon monoxide from CODH or the methyl group donated by the CFeSP protein in no particular order.[11] This is followed by migratory insertion to form an intermediate complex. CoA then binds to the metal and the final product, acetyl-CoA, is formed.[2][8] Some criticisms of this mechanism are that it is unbalanced in terms of electron count and the activated Ni+1 intermediate cannot be detected with electron paramagnetic resonance. Furthermore, there is evidence of the ACS catalytic cycle without any external reducing complex, which refutes the ferrodoxin activation step.[12]
The second proposed mechanism, the diamagnetic mechanism, involves a Ni0 intermediate instead of a Ni1+. After addition of the methyl group and carbon monoxide, followed by insertion to produce the metal-acetyl complex, CoA attacks to produce the final product.[8] The order in which the carbon monoxide molecule and the methyl group bind to the nickel centre has been highly debated, but no solid evidence has demonstrated preference for one over the other. Although this mechanism is electronically balanced, the idea of a Ni0 species is highly unprecedented in biology. There has also been no solid evidence supporting the presence of a zero-valent Ni species. However, similar nickel species to ACS with a Ni0 centre have been made, so the diamagnetic mechanism is not an implausible hypothesis.[13]
References
- ↑ Lindahl, PaulA. (25 June 2004). "Acetyl-coenzyme A synthase: the case for a Nip0-based mechanism of catalysis". JBIC Journal of Biological Inorganic Chemistry. 9 (5). doi:10.1007/s00775-004-0564-x.
- 1 2 3 4 5 6 7 8 Can, Mehmet; Armstrong, Fraser A.; Ragsdale, Stephen W. (23 April 2014). "Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase". Chemical Reviews. 114 (8): 4149–4174. doi:10.1021/cr400461p.
- 1 2 Hegg, Eric L. (October 2004). "Unraveling the Structure and Mechanism of Acetyl-Coenzyme A Synthase". Accounts of Chemical Research. 37 (10): 775–783. doi:10.1021/ar040002e. PMID 15491124.
- ↑ Riordan, C.G. (24 June 2004). "Synthetic chemistry and chemical precedents for understanding the structure and function of acetyl coenzyme A synthase". JBIC Journal of Biological Inorganic Chemistry. 9 (5). doi:10.1007/s00775-004-0567-7.
- 1 2 Ragsdale, Stephen W.; Kumar, Manoj (January 1996). "Nickel-Containing Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase". Chemical Reviews. 96 (7): 2515–2540. doi:10.1021/cr950058.
- 1 2 Doukov, TI; Iverson, TM; Seravalli, J; Ragsdale, SW; Drennan, CL (18 October 2002). "A Ni-Fe-Cu center in a bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase.". Science. 298 (5593): 567–72. doi:10.1126/science.1075843. PMID 12386327.
- 1 2 Drennan, CatherineL.; Doukov, TzankoI.; Ragsdale, StephenW. (18 June 2004). "The metalloclusters of carbon monoxide dehydrogenase/acetyl-CoA synthase: a story in pictures". JBIC Journal of Biological Inorganic Chemistry. 9 (5). doi:10.1007/s00775-004-0563-y.
- 1 2 3 4 Evans, David J. (2005). "Chemistry relating to the nickel enzymes CODH and ACS". Coordination Chemistry Reviews. 249 (15-16): 1582–1595. doi:10.1016/j.ccr.2004.09.012.
- ↑ Kung, Y.; Drennan, C. L. (April 2011). "A role for nickel–iron cofactors in biological carbon monoxide and carbon dioxide utilization". Current Opinion in Chemical Biology. 15 (2): 276–283. doi:10.1016/j.cbpa.2010.11.005.
- 1 2 Boer, J. L.; Mulrooney, S. B.; Hausinger, R. P. (February 2014). "Nickel-dependent metalloenzymes". Archives of Biochemistry and Biophysics. 544: 142–152. doi:10.1016/j.abb.2013.09.002. PMC 3946514
 . PMID 24036122.
. PMID 24036122. - 1 2 Seravalli, J.; Ragsdale, S. W. (18 January 2008). "Pulse-Chase Studies of the Synthesis of Acetyl-CoA by Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase: EVIDENCE FOR A RANDOM MECHANISM OF METHYL AND CARBONYL ADDITION". Journal of Biological Chemistry. 283 (13): 8384–8394. doi:10.1074/jbc.M709470200.
- ↑ Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O. (2006). Nickel and its surprising impact in nature. Chichester, West Sussex, England: Wiley. pp. 377–380. ISBN 9780470016718.
- ↑ Lindahl, Paul A. (25 June 2004). "Acetyl-coenzyme A synthase: the case for a Ni0-based mechanism of catalysis". Journal of Biological Inorganic Chemistry. 9 (5). doi:10.1007/s00775-004-0564-x.