Acromegaly
| Acromegaly | |
|---|---|
|
Facial features of a person with acromegaly. The cheekbones are pronounced, the forehead bulges, the jaw is enlarged, and facial lines prominent. | |
| Classification and external resources | |
| Pronunciation | /ˌækrəˈmɛɡəli, -roʊ-/;[1][2] |
| Specialty | Endocrinology |
| ICD-10 | E22.0inusl |
| ICD-9-CM | 253.0 |
| OMIM | 102200 |
| DiseasesDB | 114 |
| MedlinePlus | 000321 |
| eMedicine | med/27 derm/593 |
| Patient UK | Acromegaly |
| MeSH | D000172 |
| Orphanet | 963 |

Acromegaly is a condition that results from excess growth hormone (GH) after the growth plates have closed.[3] The initial symptom is typically enlargement of the hands and feet. There may also be enlargement of the forehead, jaw, and nose. Other symptoms may include joint pain, thicker skin, deepening of the voice, headaches, and problems with vision. Complications of the disease may include type 2 diabetes, sleep apnea, and high blood pressure.[3]
Acromegaly is typically due to the pituitary gland producing too much growth hormone. In more than 95% of people the excess production is due to a benign tumor, known as a pituitary adenoma. The condition is not inherited from a person's parents. Rarely acromegally is due to tumors in other parts of the body. Diagnosis is by measuring growth hormone after a person has drunk glucose or measuring insulin-like growth factor I in the blood. After diagnosis medical imaging of the pituitary is carried out to look for an adenoma. If excess growth hormone is produced during childhood the result is gigantism.[3]
Treatment option include surgery to remove the tumor, medications, and radiation therapy. Surgery is usually the preferred treatment and is most effective when the tumor is smaller. In those in whom surgery is not effective, medications of the somatostatin analogue or GH receptor antagonist type may be used. The effects of radiation therapy are more gradual than that of surgery or medication.[3] Without treatment those affected live on average 10 years less; however, with treatment life expectancy is typically normal.[4]
Acromegaly affects about 6 per 100,000 people. It is most commonly diagnosed in middle age.[3] Males and females are affected equally frequently.[5] The first medical description of the disorder occurred in 1772 by Nicolas Saucerotte.[6][7] The term is from Greek ἄκρον akron meaning "extremity" and μέγα mega meaning "large".[3]
Signs and symptoms
Features that result from high level of GH or expanding tumor include:
- Soft tissue swelling visibly resulting in enlargement of the hands, feet, nose, lips and ears, and a general thickening of the skin
- Soft tissue swelling of internal organs, notably the heart with attendant weakening of its muscularity, and the kidneys, also the vocal cords resulting in a characteristic thick, deep voice and slowing of speech
- Generalized expansion of the skull at the fontanelle
- Pronounced brow protrusion, often with ocular distension (frontal bossing)
- Pronounced lower jaw protrusion (prognathism) with attendant macroglossia (enlargement of the tongue) and teeth spacing
- Hypertrichosis, hyperpigmentation and hyperhidrosis may occur in these patients.[8]:499
- Acrochordon (skin tags)
- Carpal tunnel syndrome
-
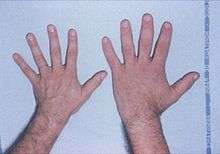
Compared with the hand of a typical person (left), the hand of a patient with acromegaly (right) is enlarged, with fingers that are widened, thickened and stubby, and with thicker soft tissue
-
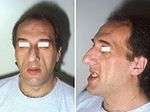
Mandibular overgrowth leads to prognathism, maxillary widening, teeth spacing and malocclusion.
-
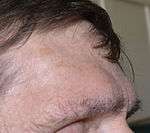
Brow ridge and forehead protrusion remaining after tumor removal and tissue swelling eliminated
-

Lower jaw showing the classic spacing of teeth due to acromegaly
Complications
- Severe headache
- Arthritis and carpal tunnel syndrome
- Enlarged heart
- Liver fibrosis and bile duct hyperplasia.
- Hypertension
- Diabetes mellitus (excess of GH leads to insulin resistance)
- Heart failure
- Kidney failure
- Colorectal cancer [9]
- Compression of the optic chiasm leading to loss of vision in the outer visual fields (typically bitemporal hemianopia.)
- Increased palmar sweating and sebum production over the face (seborrhea) are clinical indicators of active GH-producing pituitary tumors. These symptoms can also be used to monitor the activity of the tumor after surgery, although biochemical monitoring is confirmatory.
Causes
Pituitary adenoma
About 98% of cases of acromegaly are due to the overproduction of growth hormone by a benign tumor of the pituitary gland called an adenoma.[10] These tumors produce excessive growth hormone and compress surrounding brain tissues as they grow larger. In some cases, they may compress the optic nerves. Expansion of the tumor may cause headaches and visual disturbances. In addition, compression of the surrounding normal pituitary tissue can alter production of other hormones, leading to changes in menstruation and breast discharge in women and impotence in men because of reduced testosterone production.
A marked variation in rates of GH production and the aggressiveness of the tumor occurs. Some adenomas grow slowly and symptoms of GH excess are often not noticed for many years. Other adenomas grow rapidly and invade surrounding brain areas or the sinuses, which are located near the pituitary. In general, younger patients tend to have more aggressive tumors.
Most pituitary tumors arise spontaneously and are not genetically inherited. Many pituitary tumors arise from a genetic alteration in a single pituitary cell which leads to increased cell division and tumor formation. This genetic change, or mutation, is not present at birth, but is acquired during life. The mutation occurs in a gene that regulates the transmission of chemical signals within pituitary cells; it permanently switches on the signal that tells the cell to divide and secrete growth hormones. The events within the cell that cause disordered pituitary cell growth and GH oversecretion currently are the subject of intensive research.
Pituitary adenomas and diffuse somatomammotroph hyperplasia may result from somatic activating mutations GNAS, which may be acquired or associated with McCune-Albright syndrome.[11][12]
Other tumors
In a few patients, acromegaly is caused not by pituitary tumors, but by tumors of the pancreas, lungs, and adrenal glands. These tumors also lead to an excess of GH, either because they produce GH themselves or, more frequently, because they produce GHRH (growth hormone releasing hormone), the hormone that stimulates the pituitary to make GH. In these patients, the excess GHRH can be measured in the blood and establishes that the cause of the acromegaly is not due to a pituitary defect. When these nonpituitary tumors are surgically removed, GH levels fall and the symptoms of acromegaly improve.
In patients with GHRH-producing, non-pituitary tumors, the pituitary still may be enlarged and may be mistaken for a tumor. Therefore, it is important that physicians carefully analyze all "pituitary tumors" removed from patients with acromegaly so as to not overlook the possibility that a tumor elsewhere in the hypoglycoma is causing the disorder.
Diagnosis

If acromegaly is suspected, medical imaging and medical laboratory investigations are generally used together to confirm or rule out the presence of this condition.
IGF1 provides the most sensitive lab test for the diagnosis of acromegaly, and a GH suppression test following an oral glucose load, which is a very specific lab test, will confirm the diagnosis following a positive screening test for IGF1. A single value of the GH is not useful in view of its pulsatality (levels in the blood vary greatly even in healthy individuals).
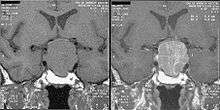
GH levels taken 2 hours after a 75- or 100-gram glucose tolerance test are helpful in the diagnosis: GH levels are suppressed below 1 μg/l in normal people, and levels higher than this cutoff are confirmatory of acromegaly.
Other pituitary hormones must be assessed to address the secretory effects of the tumor, as well as the mass effect of the tumor on the normal pituitary gland. They include thyroid stimulating hormone (TSH), gonadotropic hormones (FSH, LH), adrenocorticotropic hormone, and prolactin.
An MRI of the brain focusing on the sella turcica after gadolinium administration allows for clear delineation of the pituitary and the hypothalamus and the location of the tumor. A number of other overgrowth syndromes can result in similar problems.
Pseudoacromegaly
Pseudoacromegaly is a condition with the usual acromegaloid features, but without an increase in growth hormone and IGF-1. It is frequently associated with insulin resistance.[13] Cases have been reported due to minoxidil at an unusually high dose.[14] It can also be caused by a selective postreceptor defect of insulin signalling, leading to the impairment of metabolic, but preservation of mitogenic, signalling.[15]
Treatment
The goals of treatment are to reduce GH production to normal levels, to relieve the pressure that the growing pituitary tumor exerts on the surrounding brain areas, to preserve normal pituitary function, and to reverse or ameliorate the symptoms of acromegaly. Currently, treatment options include surgical removal of the tumor, drug therapy, and radiation therapy of the pituitary.
Medications
Somatostatin analogues
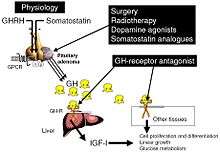
The primary current medical treatment of acromegaly is to use somatostatin analogues – octreotide (Sandostatin) or lanreotide (Somatuline). These somatostatin analogues are synthetic forms of a brain hormone, somatostatin, which stops GH production. The long-acting forms of these drugs must be injected every 2 to 4 weeks for effective treatment. Most patients with acromegaly respond to this medication. In many patients, GH levels fall within one hour and headaches improve within minutes after the injection. Octreotide and lanreotide are effective for long-term treatment. Octreotide and lanreotide have also been used successfully to treat patients with acromegaly caused by non-pituitary tumors.
Somatostatin analogues are also sometimes used to shrink large tumors before surgery.
Because octreotide inhibits gastrointestinal and pancreatic function, long-term use causes digestive problems such as loose stools, nausea, and gas in one third of patients. In addition, approximately 25 percent of patients develop gallstones, which are usually asymptomatic.[16] In some cases, octreotide treatment can cause diabetes due to the fact that somatostatin and its analogues can inhibit the release of insulin. On the other hand, scientists have found that in some acromegaly patients who already have diabetes, octreotide can reduce the need for insulin and improve blood sugar control.
Dopamine agonists
For those who are unresponsive to somatostatin analogues, or for whom they are otherwise contraindicated, it is possible to treat using one of the dopamine agonists, bromocriptine or cabergoline. As tablets rather than injections, they cost considerably less. These drugs can also be used as an adjunct to somatostatin analogue therapy. They are most effective in those whose pituitary tumours cosecrete prolactin. Side effects of these dopamine agonists include gastrointestinal upset, nausea, vomiting, light-headedness when standing, and nasal congestion. These side effects can be reduced or eliminated if medication is started at a very low dose at bedtime, taken with food, and gradually increased to the full therapeutic dose. However, bromocriptine lowers GH and IGF-1 levels and reduces tumor size in fewer than half of patients with acromegaly. Some patients report improvement in their symptoms although their GH and IGF-1 levels still are elevated.
Growth hormone receptor antagonists
The latest development in the medical treatment of acromegaly is the use of growth hormone receptor antagonists. The only available member of this family is pegvisomant (Somavert). By blocking the action of the endogenous growth hormone molecules, this compound is able to control disease activity of acromegaly in virtually all patients. Pegvisomant has to be administered subcutaneously by daily injections. Combinations of long-acting somatostatin analogues and weekly injections of pegvisomant seem to be equally effective as daily injections of pegvisomant.
Surgery
Surgery is a rapid and effective treatment, of which there are two alternative methods. The first method, a procedure known as endonasal transphenoidal surgery, involves the surgeon reaching the pituitary through an incision in the nasal cavity wall. The wall is reached by passing through the nostrils with microsurgical instruments. The second method is transphenoidal surgery during which an incision is made into the gum beneath the upper lip. Further incisions are made to cut through the septum to reach the nasal cavity, where the pituitary is located. Endonasal transphenoidal surgery is a less invasive procedure with a shorter recovery time than the older method of transphenoidal surgery, and the likelihood of removing the entire tumor is greater with reduced side effects. Consequently, endonasal transphenoidal surgery is often used as a first option, with transphenoidal and other treatments, such as medicinal therapy or stereotactic radiosurgery, used to reduce the remaining adverse effects of the remaining tumor.
These procedures normally relieve the pressure on the surrounding brain regions and lead to a lowering of GH levels. Surgery is most successful in patients with blood GH levels below 40 ng/ml before the operation and with pituitary tumors no larger than 10 mm in diameter. Success depends on the skill and experience of the surgeon. The success rate also depends on what level of GH is defined as a cure. The best measure of surgical success is normalization of GH and IGF-1 levels. Ideally, GH should be less than 2 ng/ml after an oral glucose load. A review of GH levels in 1,360 patients worldwide immediately after surgery revealed that 60% had random GH levels below 5 ng/ml. Complications of surgery may include cerebrospinal fluid leaks, meningitis, or damage to the surrounding normal pituitary tissue, requiring lifelong pituitary hormone replacement.
Even when surgery is successful and hormone levels return to normal, patients must be carefully monitored for years for possible recurrence. More commonly, hormone levels may improve, but not return completely to normal. These patients may then require additional treatment, usually with medications.
Radiation therapy
Radiation therapy has been used both as a primary treatment and combined with surgery or drugs. It is usually reserved for patients who have tumor remaining after surgery. These patients often also receive medication to lower GH levels. Radiation therapy is given in divided doses over four to six weeks. This treatment lowers GH levels by about 50 percent over 2 to 5 years. Patients monitored for more than 5 years show significant further improvement. Radiation therapy causes a gradual loss of production of other pituitary hormones with time. Loss of vision and brain injury, which have been reported, are very rare complications of radiation treatments.
Choices
No single treatment is effective for all patients. Treatment should be individualized depending on patient characteristics, such as age and tumor size. If the tumor has not yet invaded surrounding brain tissues, removal of the pituitary adenoma by an experienced neurosurgeon is usually the first choice. After surgery, a patient must be monitored for a long time for increasing GH levels. If surgery does not normalize hormone levels or a relapse occurs, a doctor will usually begin additional drug therapy. The current first choice is generally octreotide or lanreotide. However, bromocriptine or cabergoline are much cheaper and easier to administer. With both types of medication, long-term therapy is necessary because their withdrawal can lead to rising GH levels and tumor re-expansion. Radiation therapy is generally used for patients whose tumors are not completely removed by surgery; for patients who are not good candidates for surgery because of other health problems; and for patients who do not respond adequately to surgery and medication.
Prognosis
Upon successful treatment, symptoms and complications generally improve substantially or disappear, including headaches, visual disturbances, excess sweating, and diabetes.[17] Soft-tissue swellings generally decrease and acromegaly-associated facial features gradually return towards normal, although this may take some time.[17] Life expectancy after the successful treatment of early acromegaly is equal to that of the normal population.[17]
Notable cases
Famous people with acromegaly include:
- Richard Kiel, actor, "Jaws" from two James Bond movies and Mr. Larson in Happy Gilmore[18] He grew up to 7 ft 1½ in, but was slightly under 7 ft in his later years due to age and injury.
- Pío Pico, the last Mexican Governor of California (1801–1894), manifested acromegaly without gigantism between at least 1847 and 1858. Some time after 1858, signs of the growth hormone-producing tumor disappeared along with all the secondary effects the tumor had caused in him. He looked normal in his 90s.[19] His remarkable recovery is likely an example of spontaneous selective pituitary tumor apoplexy.[20]
- Adam Rainer, born in 1899, an Austrian man, is so far the only person in recorded history to have been both a dwarf and a giant. His rapid change from dwarf to giant is believed to have been caused by acromegaly.[21] He died on 4 March 1950.
- Anthony Robbins, motivational speaker[22]
- André the Giant (born André Rousimoff), French professional wrestler and actor[23][24] was 2.13 m (7 ft) tall after back surgery; his original wrestling statistics listed him at 2.23 m (7 ft 4 in). He died at the age of 46 and weighed 520 pounds (240 kg). (He chose not to be treated and died from cardiac complications of acromegaly.)
- Big Show (born Paul Wight), American professional wrestler and actor, currently working for the WWE, had his pituitary tumor removed in 1991.[25]
- The Great Khali (born Dalip Singh Rana), Indian professional wrestler, is best known for his tenure with WWE. He had his pituitary tumor removed in 2012, aged 39.[26]
- Antônio Silva Brazilian mixed martial artist, competes in the heavyweight division of the UFC.
- Maurice Tillet (1903-1954), Russian-born French professional wrestler, is better known by his ring name, the French Angel.[27]
- Carel Struycken, Dutch actor, 2.13 m (7 ft), is best known for playing Lurch in The Addams Family film trilogy.[28]
- Irwin Keyes American Actor. Best known for portraying Hugo Mojoloweski, George's occasional bodyguard on The Jeffersons[29]
- Paul Benedict American Actor. Best known for portraying Harry Bentley, The Jeffersons English next door neighbour[30]
- Rondo Hatton American Actor. A Hollywood favorite in B-movie horror films of the 1930s and 1940s. Hatton's disfigurement, due to acromegaly, developed over time, beginning during his service in World War I.[31]
References
- ↑ "Acromegaly". Oxford Dictionaries. Oxford University Press. Retrieved 2016-01-20.
- ↑ "Acromegaly". Merriam-Webster Dictionary.
- 1 2 3 4 5 6 "Acromegaly". NIDDK. April 2012. Retrieved 20 August 2016.
- ↑ Ho, Ken (2011). Growth Hormone Related Diseases and Therapy: A Molecular and Physiological Perspective for the Clinician. Springer Science & Business Media. p. 400. ISBN 9781607613176.
- ↑ Pack, Allan I. (2016). Sleep Apnea: Pathogenesis, Diagnosis and Treatment (2 ed.). CRC Press. p. 291. ISBN 9781420020885.
- ↑ Pearce, John M. S. (2003). Fragments of Neurological History. World Scientific. p. 501. ISBN 9781783261109.
- ↑ Pearce, JM (September 2006). "Nicolas Saucerotte: Acromegaly before Pierre Marie.". Journal of the history of the neurosciences. 15 (3): 269–75. PMID 16887764.
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- ↑ Renehan AG, O'Connell J, O'Halloran D, et al. (2003). "Acromegaly and colorectal cancer: a comprehensive review of epidemiology, biological mechanisms, and clinical implications". Horm. Metab. Res. 35 (11-12): 712–25. doi:10.1055/s-2004-814150. PMID 14710350.
- ↑ Kasper, Dennis; Fauci, Anthony; Hauser, Stephen; Longo, Dan; J. Jameson; Loscalzo, Joseph. (April 8, 2015). Harrison's Principles of Internal Medicine (19th ed.). McGraw Hill. pp. 2269–2271. ISBN 0071802150.
- ↑ Vortmeyer, Alexander O.; Gläsker, Sven; Mehta, Gautam U.; Abu-Asab, Mones S.; Smith, Jonathan H.; Zhuang, Zhengping; Collins, Michael T.; Oldfield, Edward H. (2012-07-01). "Somatic GNAS mutation causes widespread and diffuse pituitary disease in acromegalic patients with McCune-Albright syndrome". The Journal of Clinical Endocrinology and Metabolism. 97 (7): 2404–2413. doi:10.1210/jc.2012-1274. ISSN 1945-7197. PMC 3791436
 . PMID 22564667.
. PMID 22564667. - ↑ Salenave, Sylvie; Boyce, Alison M.; Collins, Michael T.; Chanson, Philippe (2014-06-01). "Acromegaly and McCune-Albright syndrome". The Journal of Clinical Endocrinology and Metabolism. 99 (6): 1955–1969. doi:10.1210/jc.2013-3826. ISSN 1945-7197. PMC 4037730. PMID 24517150.
- ↑ Yaqub A, Yaqub N (2008). "Insulin-mediated pseudoacromegaly: a case report and review of the literature". W V Med J. 104 (5): 12–5. PMID 18846753.
- ↑ Nguyen KH, Marks JG (June 2003). "Pseudoacromegaly induced by the long-term use of minoxidil". J. Am. Acad. Dermatol. 48 (6): 962–5. doi:10.1067/mjd.2003.325. PMID 12789195.
- ↑ "Insulin-mediated "pseudoacromegaly".". Retrieved 2013-05-12.
- ↑ http://www.drugs.com/sfx/octreotide-side-effects.html
- 1 2 3 Acromegaly - Patient information Archived July 21, 2011, at the Wayback Machine. from Centre for Diabetes and Endocrinology at The Royal Berkshire NHS Foundation Trust. January 2011
- ↑ "Richard "Jaws" Kiel, Famed Bond Movie Villain, Raises Awareness Oflife-Threatening Hormone Disorder". Prnewswire.com. Retrieved 2010-07-26.
- ↑ Login IS, Login J (July 2008). "Governor Pio Pico, the monster of California...no more: lessons in neuroendocrinology". Pituitary. 13 (1): 80–6. doi:10.1007/s11102-008-0127-1. PMC 2807602. PMID 18597174. Open Access; http://www.springerlink.com/content/u7645787h2435373/fulltext.pdf
- ↑ Nawar RN, AbdelMannan D, Selman WR, Arafah BM (2008). "Pituitary tumor apoplexy: a review". J Intensive Care Med. 23 (2): 75–90. doi:10.1177/0885066607312992. PMID 18372348.
- ↑ http://thechirurgeonsapprentice.com/2015/01/20/worlds-littlest-giant-the-curious-case-of-adam-rainer/
- ↑ Plaskin, Glenn (2013-08-13). "Playboy Interview: Tony Robbins". Playboy. Retrieved 5 March 2015.
- ↑ "Andre the Giant: Bio". WWE.com. Archived from the original on 2008-01-10. Retrieved 2007-10-16.
- ↑ "André the Giant official website". Archived from the original on 4 July 2007. Retrieved 2007-07-08.
- ↑ "Big Show at home in current role", by Tim Baines, Ottawa Sun
- ↑ "WWE Star Great Khali's Growth-Inducing Tumor Removed", by Alon Harish, ABC News
- ↑ " The French Angel was more man than monster", by Greg Oliver, Producer, SLAM! Wrestling
- ↑ Dennis McLellan for the Los Angeles Times. December 27, 1992 Shedding Light on a Rare Disease : Woman Hopes O.C. Group Will Increase Awareness of Life-Threatening Effects of Excessive-Growth Illness
- ↑ Dagan, Carmel (8 July 2015). "Irwin Keyes, Horror Movie Character Actor, Dies at 63". Variety.
- ↑ Times Staff And Wire Reports (Dec 5, 2008). "Paul Benedict dies at 70; actor from 'The Jeffersons' and 'Sesame Street'". latimes.com. Los Angeles Times.
- ↑ Duryea, Bill (June 27, 1999). "Floridian: In love with a monster". St Petersburg Times.
External links
- Acromegaly at DMOZ
- 2011 American Association of CLinical Endocrinologists Guideline
- Endocrine and Metabolic Diseases Information Service