Electrophilic fluorination
Electrophilic fluorination is the combination of a carbon-centered nucleophile with an electrophilic source of fluorine to afford organofluorine compounds. Although elemental fluorine and reagents incorporating an oxygen-fluorine bond can be used for this purpose, they have largely been replaced by reagents containing a nitrogen-fluorine bond.[1]
Electrophilic fluorination offers an alternative to nucleophilic fluorination methods employing alkali or ammonium fluorides and methods employing sulfur fluorides for the preparation of organofluorine compounds. Development of electrophilic fluorination reagents has always focused on removing electron density from the atom attached to fluorine; however, compounds containing nitrogen-fluorine bonds have proven to be the most economical, stable, and safe electrophilic fluorinating agents. Electrophilic N-F reagents are either neutral or cationic and may possess either sp2- or sp3-hybridized nitrogen. Although the precise mechanism of electrophilic fluorination is currently unclear, highly efficient and stereoselective methods have been developed.
The most common fluorinating agents used for organic synthesis are N-fluoro-o-benzenedisulfonimide (NFOBS), N-fluorobenzenesulfonimide (NFSI), and Selectfluor.[1]
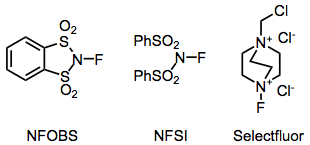
Mechanism and stereochemistry
Prevailing mechanism
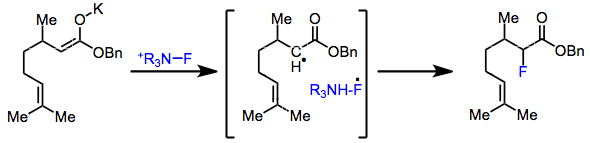
The mechanism of electrophilic fluorination remains controversial. At issue is whether the reaction proceeds via an SN2 or single-electron transfer (SET) process. In support of the SN2 mechanism, aryl Grignard reagents and aryllithiums give similar yields of fluorobenzene in combination with N-fluoro-o-benzenedisulfonimide (NFOBS), even though the tendencies of these reagents to participate in SET processes differ substantially.[2] Additionally, radical probe experiments with 5-hexenyl and cyclopropyl enol ethers did not give any rearranged products.[3]
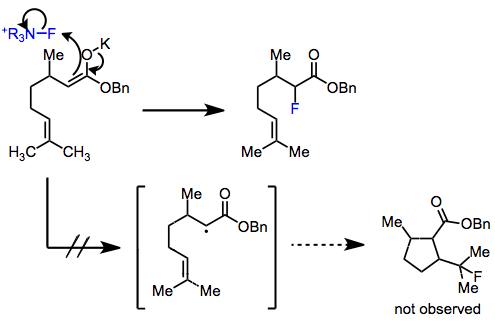
On the other hand, the lifetime of radicals in the SET process is predicted to be four orders of magnitude shorter than the detection limit of even the most sensitive of radical probes. It has been postulated that after electron transfer, immediate recombination of the fluorine radical with the alkyl radical takes place.[4]
Stereoselective variants
Stereoselective fluorinations may be either diastereoselective or enantioselective. Diastereoselective methods have focused on the use of chiral auxiliaries on the nucleophilic substrate. For fluorinations of carbonyl compounds, chiral oxazolidinones have been used with success.[5]
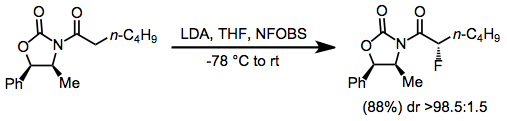
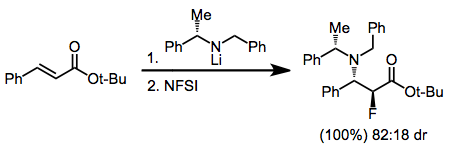
Tandem conjugate addition incorporating a chiral nucleophile has been used to synthesize β-amino α-fluoro esters in chiral, non-racemic form.
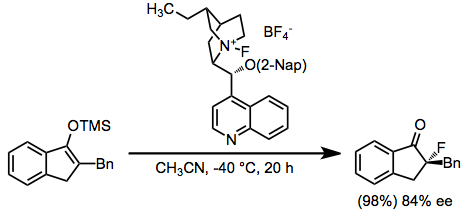
Enantioselective methods employ stoichiometric amounts of chiral fluorinating agents. N-fluoroammonium salts of cinchona alkaloids represent the state of the art for reactions of this type. In addition, these reagents are easily synthesized from Selectfluor and the parent alkaloids.[6]
Scope and limitations
Fluorinating reagents
Electrophilic N-F fluorinating reagents incorporate electron-withdrawing groups attached to nitrogen to decrease the electron density on fluorine. Although N-fluorosulfonamides are fairly weak fluorinating reagents, N-fluorosulfonimides, such as N-fluorobenzenesulfonimide (NFSI), are very effective and in common use. N-fluoro-o-benzenedisulfonimide (NFOBS) is synthesized from the disulfonic acid.[2]

The use of salts of cationic nitrogen increases the rates and yields of electrophilic fluorination, because the cationic nitrogen removes electron density from fluorine. N-fluoropyridinium ions and iminium ions can also be used as electrophilic fluorinating reagents. The counteranions of these salts, although they are not directly involved in the transfer of fluorine to the substrate, influence reactivity in subtle ways and may be adjusted using a variety of methods.[7]

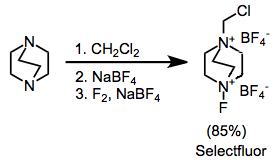
The most synthetically useful ammonium salts are the substituted DABCO bis(ammonium) ions, including Selectfluor.[8] These can be easily synthesized by alkylation followed by fluorination. The difluoro version, which might at first seem more useful, delivers only a single fluorine atom.
More specialized electrophilic fluorinating reagents, such as neutral heterocycles containing N–F bonds,[9] are useful for the fluorination of a limited range of substrates.
Nucleophilic substrates
Simple fluorinations of alkenes often produce complex mixtures of products. However, cofluorination in the presence of a nucleophile proceeds cleanly to give vicinal alkoxyfluorides.[10] Alkynes are not fluorinated with N-F reagents. A surfactant was used to facilitate contact between aqueous Selectfluor and the alkene.

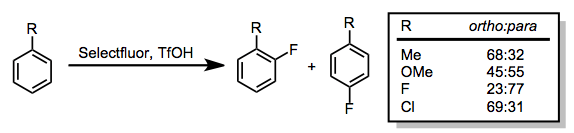
Fluorination of electron-rich aromatic compounds gives aryl fluorides. The two most common problems in this class of reactions are low ortho/para selectivities and dearomatization (the latter is a particularly significant problem for phenols).[11]
Enol ethers and glycals are nucleophilic enough to be fluorinated by Selectfluor.[12] Similar to other alkenes, cohalogenation can be accomplished either by isolation of the intermediate adduct and reaction with a nucleophile or direct displacement of DABCO in situ. Enols can be fluorinated enantioselectively (see above) in the presence of a chiral fluorinating agent.

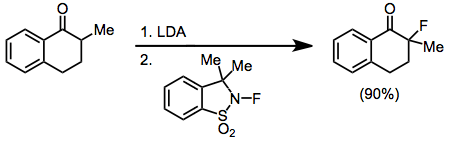
Metal enolates are compatible with many fluorinating reagents, including NFSI, NFOBS, and sulfonamides. However, the specialized reagent 2-fluoro-3,3-dimethyl-2,3-dihydrobenzo[d]isothiazole 1,1-dioxide consistently affords better yields of monofluorinated carbonyl compounds in reactions with lithium enolates. Other metal enolates afforded large amounts of difluorinated products.[13]
Comparison with other methods
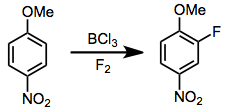
Although the use of molecular fluorine as an electrophilic fluorine source is often the cheapest and most direct method, F2 often forms radicals and reacts with C-H bonds without selectivity. Proton sources or Lewis acids are required to suppress radical formation, and even when these reagents are present, only certain substrates react with high selectivity.[14] Handling toxic, gaseous F2 requires special apparatus and great care to avoid contact with air or water.
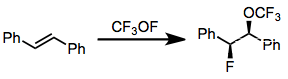
Reagents containing O-F bonds, such as CF3OF, tend to be more selective for monofluorination than N-F reagents.[15] However, difficulties associated with handling and their extreme oxidizing power have led to their replacement with N-F reagents.
Xenon di-, tetra-, and hexafluoride are selective monofluorinating reagents. However, their instability and high cost have made them less popular than organic fluorinating agents.[16]
Experimental conditions and procedure
Typical conditions
Although fluorinations employing N-F reagents do not use molecular fluorine directly, they are almost universally prepared from F2. Proper handling of F2 requires great care and special apparatus.[17] Poly(tetrafluoroethylene) (PTFE, also known as Teflon) reaction vessels are used in preference to stainless steel or glass for reactions involving molecular fluorine. F2 blends with N2 or He are commercially available and help control the speed of delivery of fluorine. Temperatures should be kept low, and introduction of fluorine slow, to prevent free radical reactions.
See also
References
- 1 2 Badoux, J.; Cahard, D. Org. React. 2007, 69, 347. doi:10.1002/0471264180.or069.02
- 1 2 Davis, F. A.; Han, W.; Murphy, C. K. J. Org. Chem. 1995, 60, 4730.
- ↑ Differding, E.; Rüegg, G. M. Tetrahedron Lett. 1991, 32, 3815.
- ↑ Piana, S.; Devillers, I.; Togni, A.; Rothlisberger, U. Angew. Chem. Int. Ed. Engl. 2002, 41, 979.
- ↑ Davis, F. A.; Kasu, P. V. N. Tetrahedron Lett. 1998, 39, 6135.
- ↑ Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001.
- ↑ Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K. Bull. Chem. Soc. Jpn. 1991, 64, 1081.
- ↑ Stavber, S.; Zupan, M.; Poss, A. J.; Shia, G. A. Tetrahedron Lett. 1995, 36, 6769.
- ↑ Laali, K. K.; Tanaka, M.; Forohar, F.; Cheng, M.; Fetzer, J. C. J. Fluorine Chem. 1998, 91, 185.
- ↑ Lal, G. S. J. Org. Chem. 1993, 58, 2791.
- ↑ Zupan, M.; Iskra, J.; Stavber, S. Bull. Chem. Soc. Jpn. 1995, 68, 1655.
- ↑ Albert, M.; Dax, K.; Ortner, J. Tetrahedron 1998, 54, 4839.
- ↑ Differding, E.; Lang, R. W. Helv. Chim. Acta. 1989, 72, 1248.
- ↑ Chambers, R. D.; Hutchinson, J.; Sandford, G. J. Fluorine Chem. 1999, 100, 63.
- ↑ Rozen, S. Chem. Rev. 1996, 96, 1717.
- ↑ Ramsden, C. A.; Smith, R. G. J. Am. Chem. Soc. 1998, 120, 6842.
- ↑ Umemoto, T.; Nagayoshi, M. Bull. Chem. Soc. Jpn. 1996, 69, 2287.