Heritability

Heritability is a statistic used in breeding and genetics works that estimates how much variation in a phenotypic trait in a population is due to genetic variation among individuals in that population.[1] Other causes of measured variation in a trait are characterized as environmental factors, including measurement error. In human studies of heritability these are often apportioned into factors from "shared environment" and "non-shared environment" based on whether they tend to result in persons brought up in the same household more or less similar to persons who were not.
Heritability is estimated by comparing individual phenotypic variation among related individuals in a population. Heritability is an important concept in quantitative genetics, particularly in selective breeding and behavior genetics (for instance, twin studies).
Overview
Heritability measures the fraction of phenotype variability that can be attributed to genetic variation. This is not the same as saying that this fraction of an individual phenotype is caused by genetics. In addition, heritability can change without any genetic change occurring, such as when the environment starts contributing to more variation. A case in point, consider that both genes and environment have the potential to influence intelligence. Heritability could increase if genetic variation increases, causing individuals to show more phenotypic variation, like showing different levels of intelligence. On the other hand, heritability might also increase if the environmental variation decreases, causing individuals to show less phenotypic variation, like showing more similar levels of intelligence. Heritability increases when genetics are contributing more variation or because non-genetic factors are contributing less variation; what matters is the relative contribution. Heritability is specific to a particular population in a particular environment.
The extent of dependence of phenotype on environment can also be a function of the genes involved. Matters of heritability are complicated because genes may canalize a phenotype, making its expression almost inevitable in all occurring environments. Individuals with the same genotype can also exhibit different phenotypes through a mechanism called phenotypic plasticity, which makes heritability difficult to measure in some cases. Recent insights in molecular biology have identified changes in transcriptional activity of individual genes associated with environmental changes. However, there are a large number of genes whose transcription is not affected by the environment.[2]
Estimates of heritability use statistical analyses to help to identify the causes of differences between individuals. Since heritability is concerned with variance, it is necessarily an account of the differences between individuals in a population. Heritability can be univariate – examining a single trait – or multivariate – examining the genetic and environmental associations between multiple traits at once. This allows a test of the genetic overlap between different phenotypes: for instance hair color and eye color. Environment and genetics may also interact, and heritability analyses can test for and examine these interactions (GxE models).
A prerequisite for heritability analyses is that there is some population variation to account for. This last point highlights the fact that heritability cannot take into account the effect of factors which are invariant in the population. Factors may be invariant if they are absent and do not exist in the population, such as no one having access to a particular antibiotic, or because they are omni-present, like if everyone is drinking coffee. In practice, all human behavioral traits vary and almost all traits show some heritability.[3]
Definition
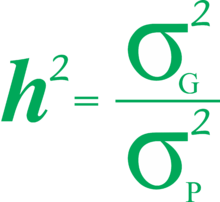
Any particular phenotype can be modeled as the sum of genetic and environmental effects:[4]
- Phenotype (P) = Genotype (G) + Environment (E).
Likewise the phenotypic variance in the trait – Var (P) – is the sum of effects as follows:
- Var(P) = Var(G) + Var(E) + 2 Cov(G,E).
In a planned experiment Cov(G,E) can be controlled and held at 0. In this case, heritability is defined as:
- .

H2 is the broad-sense heritability. This reflects all the genetic contributions to a population's phenotypic variance including additive, dominant, and epistatic (multi-genic interactions), as well as maternal and paternal effects, where individuals are directly affected by their parents' phenotype, such as with milk production in mammals.
A particularly important component of the genetic variance is the additive variance, Var(A), which is the variance due to the average effects (additive effects) of the alleles. Since each parent passes a single allele per locus to each offspring, parent-offspring resemblance depends upon the average effect of single alleles. Additive variance represents, therefore, the genetic component of variance responsible for parent-offspring resemblance. The additive genetic portion of the phenotypic variance is known as Narrow-sense heritability and is defined as

An upper case H2 is used to denote broad sense, and lower case h2 for narrow sense.
For traits which are not continuous but dichotomous such as a disease or additional toe, the contribution of the various alleles can be considered to be a sum, which past a threshold, manifests itself as the trait, giving the liability threshold model in which heritability can be estimated and selection modeled.
Additive variance is important for selection. If a selective pressure such as improving livestock is exerted, the response of the trait is directly related to narrow-sense heritability. The mean of the trait will increase in the next generation as a function of how much the mean of the selected parents differs from the mean of the population from which the selected parents were chosen. The observed response to selection leads to an estimate of the narrow-sense heritability (called realized heritability). This is the principle underlying artificial selection or breeding.
Example
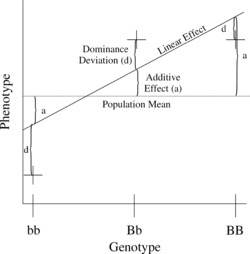
The simplest genetic model involves a single locus with two alleles (b and B) affecting one quantitative phenotype.
The number of B alleles can vary from 0, 1, or 2. For any genotype, BiBj, the expected phenotype can then be written as the sum of the overall mean, a linear effect, and a dominance deviation:
- = Population mean + Additive Effect () + Dominance Deviation ().



The additive genetic variance at this locus is the weighted average of the squares of the additive effects:

where

There is a similar relationship for variance of dominance deviations:

where

The linear regression of phenotype on genotype is shown in Figure 1.
Estimating heritability
Since only P can be observed or measured directly, heritability must be estimated from the similarities observed in subjects varying in their level of genetic or environmental similarity. The statistical analyses required to estimate the genetic and environmental components of variance depend on the sample characteristics. Briefly, better estimates are obtained using data from individuals with widely varying levels of genetic relationship - such as twins, siblings, parents and offspring, rather than from more distantly related (and therefore less similar) subjects. The standard error for heritability estimates is improved with large sample sizes.
In non-human populations it is often possible to collect information in a controlled way. For example, among farm animals it is easy to arrange for a bull to produce offspring from a large number of cows and to control environments. Such experimental control is generally not possible when gathering human data, relying on naturally occurring relationships and environments.
Studies of human heritability often utilize adoption study designs, often with identical twins who have been separated early in life and raised in different environments (see for example Fig. 2). Such individuals have identical genotypes and can be used to separate the effects of genotype and environment. A limit of this design is the common prenatal environment and the relatively low numbers of twins reared apart. A second and more common design is the twin study in which the similarity of identical and fraternal twins is used to estimate heritability. These studies can be limited by the fact that identical twins are not completely genetically identical, potentially resulting in an underestimation of heritability. Studies of twins also examine differences between twins and non-twin siblings, for instance to examine phenomena such as intrauterine competition (for example, twin-to-twin transfusion syndrome).
Heritability estimates are always relative to the genetic and environmental factors in the population, and are not absolute measurements of the contribution of genetic and environmental factors to a phenotype. Heritability estimates reflect the amount of variation in genotypic effects compared to variation in environmental effects.
Heritability can be made larger by diversifying the genetic background, e.g., by using only very out bred individuals (which increases VarG) and/or by minimizing environmental effects (decreasing VarE). The converse also holds. Due to such effects, different populations of a species might have different heritabilities for the same trait.
In observational studies, or because of evokative effects (where a genome evokes environments by its effect on them), G and E may covary: gene environment correlation. Depending on the methods used to estimate heritability, correlations between genetic factors and shared or non-shared environments may or may not be confounded with heritability.[5]
Common misunderstandings of heritability estimates
A common estimate of heritability is called the Heritability Index (HI), which ranges from 0 - 1. An HI index of 0 means that none of the variability of the trait among individuals in the study sample is the result of genetic factors; an HI of 1 indicates that all of the variability of the trait among individuals in the study sample is the result of genetic factors.
Heritability estimates are often misinterpreted if it is not understood that they refer to the proportion of variation among individuals on a trait that is the result of genetic factors. It does not indicate the degree of genetic influence on the development of a trait of an individual. For example, it is incorrect to say that since the heritability of personality traits is about .6, that means that 60% of your personality is inherited from your parents and 40% comes from the environment.
Even a highly heritable trait (such as eye color) assumes environmental inputs which are required for development: for instance temperatures and an atmosphere supporting life, etc. A more useful distinction than "nature vs. nurture" is "obligate vs. facultative"—under typical environmental ranges, what traits are more "obligate" (e.g., the nose—everyone has a nose) or more "facultative" (sensitive to environmental variations, such as specific language learned during infancy). Another useful distinction is between traits that are likely to be adaptations (such as the umbilical cord) vs. those that are byproducts of adaptations (such as the belly button), or are due to random variation (non-adaptive variation in belly button shape, e.g., convex or concave).
Estimation methods
There are essentially two schools of thought regarding estimation of heritability.
One school of thought was developed by Sewall Wright at The University of Chicago, and further popularized by C. C. Li (University of Chicago) and J. L. Lush (Iowa State University). It is based on the analysis of correlations and, by extension, regression. Path Analysis was developed by Sewall Wright as a way of estimating heritability.
The second was originally developed by R. A. Fisher and expanded at The University of Edinburgh, Iowa State University, and North Carolina State University, as well as other schools. It is based on the analysis of variance of breeding studies, using the intraclass correlation of relatives. Various methods of estimating components of variance (and, hence, heritability) from ANOVA are used in these analyses.
Regression/correlation methods of estimation
The first school of estimation uses regression and correlation to estimate heritability.
Selection experiments
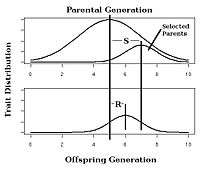
Calculating the strength of selection, S (the difference in mean trait between the population as a whole and the selected parents of the next generation, also called the selection differential[6]) and response to selection R (the difference in offspring and whole parental generation mean trait) in an artificial selection experiment will allow calculation of realized heritability as the response to selection relative to the strength of selection, h2=R/S as in Fig. 3.
Comparison of close relatives
In the comparison of relatives, we find that in general,
where r can be thought of as the coefficient of relatedness, b is the coefficient of regression and t the coefficient of correlation.

Parent-offspring regression
Heritability may be estimated by comparing parent and offspring traits (as in Fig. 4). The slope of the line (0.57) approximates the heritability of the trait when offspring values are regressed against the average trait in the parents. If only one parent's value is used then heritability is twice the slope. (Note that this is the source of the term "regression," since the offspring values always tend to regress to the mean value for the population, i.e., the slope is always less than one). This regression effect also underlies the DeFries Fulker method for analyzing twins selected for one member being affected.[7]
Sibling comparison
A basic approach to heritability can be taken using full-Sib designs: comparing similarity between siblings who share both a biological mother and a father.[8] When there is only additive gene action, this sibling phenotypic correlation is an index of familiarity – the sum of half the additive genetic variance plus full effect of the common environment. It thus places an upper-limit on additive heritability of twice the full-Sib phenotypic correlation. Half-Sib designs compare phenotypic traits of siblings that share one parent with other sibling groups.
Twin studies
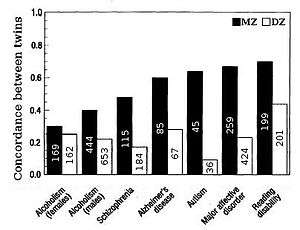
Heritability for traits in humans is most frequently estimated by comparing resemblances between twins (Fig. 2 & 5). "The advantage of twin studies, is that the total variance can be split up into genetic, shared or common environmental, and unique environmental components, enabling an accurate estimation of heritability".[9] Fraternal or dizygotic (DZ) twins on average share half their genes (assuming there is no assortative mating for the trait), and so identical or monozygotic (MZ) twins on average are twice as genetically similar as DZ twins. A crude estimate of heritability, then, is approximately twice the difference in correlation between MZ and DZ twins, i.e. Falconer's formula H2=2(r(MZ)-r(DZ)).
The effect of shared environment, c2, contributes to similarity between siblings due to the commonality of the environment they are raised in. Shared environment is approximated by the DZ correlation minus half heritability, which is the degree to which DZ twins share the same genes, c2=DZ-1/2h2. Unique environmental variance, e2, reflects the degree to which identical twins raised together are dissimilar, e2=1-r(MZ).
The methodology of the classical twin study has been criticized, but some of these criticisms do not take into account the methodological innovations and refinements described above.
Extended pedigree design
While often heritability is analyzed in single generations: comparing MZ twins raised apart, or comparing the similarity of MZ and DZ twins, considerable power can be gained using more complex relationships. By studying a trait in multi-generational families, the multiple recombination of genetic and environmental effects can be decomposed using software such as ASReml and heritability estimated.[10] This design is helpful for untangling confounds such as reverse causality, maternal effects such as the prenatal environment, and confounding of genetic dominance, shared environment, and maternal gene effects[11][12] but is generally less powerful than the twin design for obtaining heritability estimates.
Analysis of variance methods of estimation
The second set of methods of estimation of heritability involves ANOVA and estimation of variance components.
Basic model
We use the basic discussion of Kempthorne (1957 [1969]). Considering only the most basic of genetic models, we can look at the quantitative contribution of a single locus with genotype Gi as

where
is the effect of genotype Gi

and is the environmental effect.

Consider an experiment with a group of sires and their progeny from random dams. Since the progeny get half of their genes from the father and half from their (random) mother, the progeny equation is

Intraclass correlations
Consider the experiment above. We have two groups of progeny we can compare. The first is comparing the various progeny for an individual sire (called within sire group). The variance will include terms for genetic variance (since they did not all get the same genotype) and environmental variance. This is thought of as an error term.
The second group of progeny are comparisons of means of half sibs with each other (called among sire group). In addition to the error term as in the within sire groups, we have an addition term due to the differences among different means of half sibs. The intraclass correlation is
- ,

since environmental effects are independent of each other.
The ANOVA
In an experiment with sires and progeny per sire, we can calculate the following ANOVA, using as the genetic variance and as the environmental variance:




| Source | d.f. | Mean Square | Expected Mean Square |
|---|---|---|---|
| Among sire groups | |||
| Within sire groups |






The term is the intraclass correlation among half sibs. We can easily calculate . The Expected Mean Square is calculated from the relationship of the individuals (progeny within a sire are all half-sibs, for example), and an understanding of intraclass correlations.


Model with additive and dominance terms
For a model with additive and dominance terms, but not others, the equation for a single locus is

where
is the additive effect of the ith allele, is the additive effect of the jth allele, is the dominance deviation for the ijth genotype, and is the environment.


Experiments can be run with a similar setup to the one given in Table 1. Using different relationship groups, we can evaluate different intraclass correlations. Using as the additive genetic variance and as the dominance deviation variance, intraclass correlations become linear functions of these parameters. In general,


- Intraclass correlation

where and are found as

P[ alleles drawn at random from the relationship pair are identical by descent], and

P[ genotypes drawn at random from the relationship pair are identical by descent].

Some common relationships and their coefficients are given in Table 2.
| Relationship | ||
|---|---|---|
| Identical Twins | ||
| Parent-Offspring | ||
| Half Siblings | ||
| Full Siblings | ||
| First Cousins | ||
| Double First Cousins |






Larger models
When a large, complex pedigree is available for estimating heritability, the most efficient use of the data is in a restricted maximum likelihood (REML) model. The raw data will usually have three or more data points for each individual: a code for the sire, a code for the dam and one or several trait values. Different trait values may be for different traits or for different time points of measurement.
The currently popular methodology relies on high degrees of certainty over the identities of the sire and dam; it is not common to treat the sire identity probabilistically. This is not usually a problem, since the methodology is rarely applied to wild populations (although it has been used for several wild ungulate and bird populations), and sires are invariably known with a very high degree of certainty in breeding programmes. There are also algorithms that account for uncertain paternity.
The pedigrees can be viewed using programs such as Pedigree Viewer , and analyzed with programs such as ASReml, VCE , WOMBAT or BLUPF90 family's programs
Response to selection
In selective breeding of plants and animals, the expected response to selection of a trait with known narrow-sense heritability can be estimated using the breeder's equation:[13]


In this equation, the Response to Selection (R) is defined as the realized average difference between the parent generation and the next generation, and the Selection Differential (S) is defined as the average difference between the parent generation and the selected parents.
For example, imagine that a plant breeder is involved in a selective breeding project with the aim of increasing the number of kernels per ear of corn. For the sake of argument, let us assume that the average ear of corn in the parent generation has 100 kernels. Let us also assume that the selected parents produce corn with an average of 120 kernels per ear. If h2 equals 0.5, then the next generation will produce corn with an average of 0.5(120-100) = 10 additional kernels per ear. Therefore, the total number of kernels per ear of corn will equal, on average, 110.
Note that heritability in the above equation is equal to the ratio only if the genotype and the environmental noise follow Gaussian distributions .

Controversies
Heritability estimates' prominent critics, such as Steven Rose,[14] Jay Joseph,[15] and Richard Bentall, focus largely on heritability estimates in behavioral sciences and social sciences. Bentall has claimed that such heritability scores are typically calculated counterintuitively to derive numerically high scores, that heritability is misinterpreted as genetic determination, and that this alleged bias distracts from other factors that researches have found more causally important, such as childhood abuse causing later psychosis.[16][17]
The controversy over heritability estimates is largely via their basis in twin studies. The scarce success of molecular-genetic studies to corroborate such population-genetic studies' conclusions is the missing heritability problem.[18] Eric Turkheimer has argued that newer molecular methods have vindicated the conventional interpretation of twin studies,[18] although it remains mostly unclear how to explain the relations between genes and behaviors.[19] According to Turkheimer, both genes and environment are heritable, genetic contribution varies by environment, and a focus on heritability distracts from other important factors.[20] Overall, however, heritability is a concept widely applicable.[12]
See also
References
Notes
- ↑ Wray, Naomi; Visscher, Peter (2008). "Estimating Trait Heritability". Nature Education. 1 (1): 29. Retrieved 24 July 2015.
- ↑ Wills, C. (2007). "Principles of Population Genetics, 4th edition". Journal of Heredity (Book Review). 98 (4): 382–382. doi:10.1093/jhered/esm035.
- review of: Hartl, Daniel L.; Clark, Andrew G. (2007). Sunderland, MA: Sinauer and Associates. pp. xv + 652. ISBN 0-87893-308-5. Missing or empty
|title=(help)
- review of: Hartl, Daniel L.; Clark, Andrew G. (2007). Sunderland, MA: Sinauer and Associates. pp. xv + 652. ISBN 0-87893-308-5. Missing or empty
- ↑ Turkheimer, Eric (October 2000). "Three Laws of Behavior Genetics and What They Mean" (PDF). Current Directions in Psychological Science. 9 (5): 160–164. doi:10.1111/1467-8721.00084. ISSN 0963-7214. Retrieved 29 October 2013.
- ↑ Kempthorne 1957
- ↑ Cattell RB (1960). "The multiple abstract variance analysis equations and solutions: for nature–nurture research on continuous variables". Psychol Rev. 67 (6): 353–372. doi:10.1037/h0043487. PMID 13691636.
- ↑ Kempthorne 1957, p. 507; or Falconer & Mackay 1995, p. 191, for example.
- ↑ Defries, J. C.; Fulker, D. W. (September 1985). "Multiple regression analysis of twin data". Behavior Genetics. 15 (5): 467–473. doi:10.1007/BF01066239. PMID 4074272.
- ↑ Falconer, Douglas S.; Mackay, Trudy F. C. (December 1995). Introduction to Quantitative Genetics (4th ed.). Longman. ISBN 978-0582243026.
- ↑ Gielen, M., Lindsey, P.J., Derom, C., Smeets, H.J.M., Souren, N.Y., Paulussen, A.D.C., Derom, R., & Nijhuis, J.G. (2008) "Modeling Genetic and Environmental Factors to IncreaseHeritability and Ease the Identification of Candidate Genes for Birth Weight: A Twin Study". Behavioral Genetics. 38(44-54):45. DOI 10.1007/s10519-007-9170-3
- ↑ Luciano, M.; Batty, G. D.; McGilchrist, M.; Linksted, P.; Fitzpatrick, B.; Jackson, C.; Pattie, A.; Dominiczak, A. F.; Morris, A. D.; Smith, B. H. (May–June 2010). "Shared genetic aetiology between cognitive ability and cardiovascular disease risk factors: Generation Scotland's Scottish family health study". Intelligence. 38 (3): 304–313. doi:10.1016/j.intell.2010.03.002.
- ↑ Hill, W. G.; Goddard, M. E.; Visscher, P. M. (2008). MacKay, Trudy F. C., ed. "Data and Theory Point to Mainly Additive Genetic Variance for Complex Traits". PLOS Genetics. 4 (2): e1000008. doi:10.1371/journal.pgen.1000008. PMC 2265475
 . PMID 18454194.
. PMID 18454194. 
- 1 2 Visscher, P. M.; Hill, W. G.; Wray, N. R. (April 2008). "Heritability in the genomics era — concepts and misconceptions" (PDF). Nature Reviews Genetics. 9 (4): 255–266. doi:10.1038/nrg2322. PMID 18319743.
- ↑ Plomin, R., DeFries, J. C., & McClearn, G. E. (1990). Behavioral genetics. New York: Freeman.
- ↑ Rose, Steven P R (2006). "Commentary: Heritability estimates—long past their sell-by date". International Journal of Epidemiology. 35 (3): 525–527. doi:10.1093/ije/dyl064. PMID 16645027.
- ↑ Jay Joseph, The Gene Illusion (New York: Algora, 2004), esp ch 5.
- ↑ Richard P Bentall, Doctoring the Mind: Is Our Current Treatment of Mental Illness Really Any Good? (New York: New York University Press, 2009), p 123–127.
- ↑ Melanie McGrath, "Doctoring the Mind: Review", The Telegraph, 2009 Jul 5.
- 1 2 Turkheimer, Eric (2011). "Still missing". Research in Human Development. 8 (3–4): 227–241. doi:10.1080/15427609.2011.625321.
- ↑ Eric Turkheimer, "Genetic prediction", Hastings Center Report, 2015 Sep/Oct;45(S1):S32–S38.
- ↑ Jay Joseph, The Trouble with Twin Studies: A Reassessment of Twin Research in the Social and Behavioral Sciences (New York & Hove: Routledge, 2015), esp p 81 chapter summaries.
Books
- Falconer, D. S. and T. Mackay. 1996. Introduction to Quantitative Genetics, Ed. 4. Longman, Essex, England.
- Kempthorne, O (1957 [1969]) An Introduction to Genetic Statistics. John Wiley. Reprinted, 1969 by Iowa State University Press.
Further reading
- Lynch, M. & Walsh, B. 1996. Genetics and Analysis of Quantitative Traits. Sinauer Associates. ISBN 0-87893-481-2.
- Johnson, Wendy; Penke, Lars; Spinath, Frank M. (2011). "Understanding Heritability: What it is and What it is Not". European Journal of Personality. 25 (4): 287–294. doi:10.1002/per.835. ISSN 0890-2070. Archived (PDF) from the original on 2011. Retrieved 15 December 2013.
External links
- Stanford Encyclopedia of Philosophy entry on Heredity and Heritability
- Quantitative Genetics Resources website, including the two volume book by Lynch and Walsh. Free access
| Lysenkoists | |
|---|---|
| Dissidents | |
| Concepts | |
| Soviet policies | |
| |