Ion-association
In chemistry, ion-association is a chemical reaction whereby ions of opposite electrical charge come together in solution to form a distinct chemical entity. Ion-associates are classified, according to the number of ions that associate with each other, as ion-pairs, ion-triplets, etc. Ion-pairs are also classified according to the nature of the interaction as contact, solvent-shared or solvent-separated. The most important factor to determine the extent of ion-association is the dielectric constant of the solvent. Ion-associates have been characterized by means of vibrational spectroscopy.
Classification
Ion-pairs are formed when a cation and anion come together.
- An+ + Bm− ⇌ AB(n-m)+
There are three distinct types of ion-pair, depending on the extent of solvation of the two ions.
- Schematic representations of ion-pairs
-

fully solvated
-
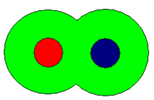
solvent-shared or solvent-separated
-
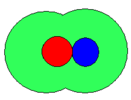
contact
In the schematic representation above, the circles represent spheres. The sizes are arbitrary and not necessarily similar as illustrated. The cation is coloured red and the anion is coloured blue. The green area represents solvent molecules in a primary solvation shell: secondary solvation is ignored. When both ions have a complete primary solvation sphere the ion-pair may be termed fully solvated. When there is about one solvent molecule between cation and anion, the ion-pair may be termed solvent-shared. Lastly when the ions are in contact with each other the ion-pair is termed a contact ion-pair. Even in a contact ion-pair, however, the ions retain most of their solvation shell. The nature of this solvation shell is generally not known with any certainty. In aqueous solution and in other donor solvents, metal cations are surrounded by between four and nine solvent molecules in the primary solvation shell,[1] but the nature of solvation of anions is mostly unknown.
An alternative name for a solvent-shared ion-pair is an outer-sphere complex. This usage is common in co-ordination chemistry and denotes a complex between a solvated metal cation and an anion. Similarly a contact ion-pair may be termed an inner-sphere complex. The essential difference between the three types is the closeness with which the ions approach each other; fully solvated > solvent-shared > contact. With fully solvated and solvent shared ion-pairs the interaction is primarily electrostatic, but in a contact ion-pair there will also be some covalent character in the bond between cation and anion.
An Ion-triplet may be formed from one cation and two anions, or from one anion and two cations.[2] Higher aggregates, such as a tetramer, (AB)4 may be formed.
Ternary ion-associates involve the association of three species.[3] Another type, named intrusion ion-pair has also been characterized.[4]
Theory
Ions of opposite charge are naturally attracted to each other by the electrostatic force.[5] This is described by Coulomb's law.

F is the force of attraction, q1 and q2 are the magnitudes of the electrical charges, ε is the dielectric constant of the medium and r is the distance between the ions. For ions in solution this is an approximation because the ions exert a polarizing effect on the solvent molecules that surround them which attenuates the electric field somewhat. Nevertheless some general conclusions can be inferred.
- Ion association will increase as
- the magnitude(s) of the electrical charge(s) q1 and q2 increase
- the magnitude of the dielectric constant, ε decreases
- the size of the ions decreases so that the distance between cation and anion, r, decreases
The equilibrium constant for ion-pair formation, K, like all equilibrium constants, is related to the standard free energy change.[6]
- .

R is the gas constant and T is the temperature in Kelvin. Free energy is made up of an enthalpy term and an entropy term.

The coulombic energy released when ions associate contributes to the enthalpy term, . In the case of contact ion-pairs, the covalent interaction energy also contributes to the enthalpy, as does the energy of displacing a solvent molecule from the solvation shell of the cation or anion. The tendency to associate is opposed by the entropy term which results from the fact that the solution containing un-associated ions is more disordered than a solution containing associates. The entropy term is similar for electrolytes of the same type, with minor differences due to solvation effects. Therefore it is the magnitude of the enthalpy term that most determines the extent of ion-association for a given electrolyte type. This explains the general rules given above.

Occurrence
Dielectric constant is the most important factor in determining the occurrence of ion-association. The following table gives some typical values.
| Solvent | dielectric constant | temperature /K |
|---|---|---|
| benzene | 2.3 | 298 |
| diethyl ether | 4.3 | 293 |
| tetrahydrofuran (THF) | 7.6 | 298 |
| dichloromethane | 9.1 | 293 |
| liquid ammonia | 17 | 273 |
| ethanol | 24.3 | 298 |
| methanol | 32.7 | 298 |
| nitromethane | 35.9 | 303 |
| dimethyl formamide (DMF) | 36.7 | 298 |
| acetonitrile | 37.5 | 293 |
| water | 78.7 | 298 |
| formamide | 109 | 293 |
Water has a relatively high dielectric constant at 25 °C, so in aqueous solutions at ambient temperatures 1:1 electrolytes such as NaCl do not form ion-pairs to an appreciable extent except when the solution is very concentrated.[7] 2:2 electrolytes (q1=2, q2=2) form ion-pairs more readily. Indeed the solvent shared ion-pair [Mg(H2O)6]2+SO42− was famously discovered to be present in seawater, in equilibrium with the contact ion-pair [Mg(H2O)5(SO4)][8] Trivalent ions such as Al3+, Fe3+ and lanthanide ions form weak complexes with monovalent anions.
The dielectric constant of water decreases with increasing temperature to ca. 55 at 100 °C and ca. 5 at the critical temperature, 217.7 °C.[9] Thus ion-pairing will become more significant in superheated water.
Solvents with a dielectric constant in the range, roughly, 20-40, show extensive ion-pair formation. For example, in acetonitrile both contact and solvent-shared ion pairs of Li(NCS) have been observed.[10] In methanol the 2:1 electrolyte Mg(NCS)2 is partially dissociated into a contact ion-pair, [Mg(NCS)]+ and the thiocyanate ion.[11]
The dielectric constant of liquid ammonia decreases from 26 at its freezing point, −80 °C to 17 at 20 °C (under pressure). Many simple 1:1 electrolytes form contact ion-pairs at ambient temperatures. The extent of ion-pairing decreases as temperature decreases. With lithium salts there is evidence to show that both inner-sphere and outer-sphere complexes exist in liquid ammonia solutions.[12]
Of the solvents with dielectric constant of 10 or less, tetrahydrofuran is particularly relevant in this context as it solvates cations strongly with the result that simple electrolytes have sufficient solubility to make the study of ion-association possible. In this solvent ion association is the rule rather than the exception. Indeed, higher associates such as tetramers are often formed.[13] Triple cations and triple anions have also been characterized in THF solutions.[14]
Ion-association is an important factor in phase-transfer catalysis since a species such as R4P+Cl− is formally neutral and so can dissolve easily in a non-polar solvent of low dielectric constant. In this case it also helps that the surface of the cation is hydrophobic.
In SN1 reactions the carbocation intermediate may form an ion-pair with an anion, particularly in solvents of low dielectric constant, such as diethylether.[15] This can affect both the kinetics of the reaction and the stereochemistry of the reaction products.
Characterization
Vibrational spectroscopy provides the most widely used means for characterizing ion-associates. Both infrared spectroscopy and Raman spectroscopy have been used. Anions containing a CN group, such as cyanide, cyanate and thiocyanide have a vibration frequency a little above 2000 cm−1 which can be easily observed as the spectra of most solvents (other than nitriles) are weak in this region. The anion vibration frequency is "shifted" on formation of ion-pairs and other associates and the extent of the shift gives information about the nature of the species. Other monovalent anions which have been studied include nitrate, nitrite and azide. Ion-pairs of monatomic anions, such as halide ions, cannot be studied by this technique. NMR spectroscopy is not very useful as association/dissociation reactions tend to be fast on the NMR time scale, giving time-averaged signals of the cation and/or anion.
Nearly the same shift of vibration frequency is observed for solvent-shared ion-pairs of LiCN, Be(CN)2 and Al(CN)3 in liquid ammonia. The extent of this type of ion-pairing decreases as the size of the cation increases. Thus, solvent-shared ion-pairs are characterized by a rather small shift of vibration frequency with respect to the "free" solvated anion and the value of the shift is not strongly dependent on the nature of the cation. The shift for contact ion-pairs is, by contrast, strongly dependent on the nature of the cation and decreases linearly with cations's charge:radius squared ratio in the two series Cs+ > Rb+ > K+ > Na+ > Li+, and Ba2+ > Sr2+ > Ca2+.[12]
The extent of contact ion-pairing can be estimated from the relative intensities of the bands due to the ion-pair and free ion. It is greater with the larger cations .[12] This is counter to the trend expected if coulombic energy were the determining factor. Instead, the formation of a contact ion-pair is seen to depend more on the energy needed to displace a solvent molecule from the primary solvation sphere of the cation. This energy decreases with the size of the cation, making ion-pairing occur to a greater extent with the larger cations. The trend may be different in other solvents.[12]
Higher ion-aggregates, sometimes triples M+X−M+, sometimes dimers of ion pairs (M+X−)2, or even larger species can be identified in the Raman spectra of some liquid ammonia solutions of Na+ salts by the presence of bands which cannot be attributed to either contact- or solvent-shared ion-pairs.[12]
Evidence for the existence of fully solvated ion-pairs in solution is mostly indirect as the spectroscopic properties of such ion-pairs are indistinguishable from those of the individual ions. Much of the evidence is based on the interpretation of conductivity measurements.[16][17]
See also
References
- ↑ Burgess, J. (1978). Metal Ions in Solution. Ellis Horwood. ISBN 0-85312-027-7. Chapter 5, "Solvation numbers".
- ↑ Fuoss, R.M.; Kraus, C.A. (1935). "Properties of Electrolytic Solutions. XV. Thermodynamic Properties of Very Weak Electrolytes". J. Amer. Chem. Soc. 57: 1–4. doi:10.1021/ja01304a001.
- ↑ Alexandrov, A.; Kostova, S. (1984). "Extraction-spectrophotometric and radiometric investigation of the ternary ion-association complex of niobium(V) with pyrocatechol and triphenyl-tetrazolium chloride". Journal of Radioanalytical and Nuclear Chemistry. Springer. 83 (2): 247–255. doi:10.1007/BF02037138.
- ↑ Fletcher, R.J.; Gans, P.; Gill, J.B.; Geyer, C. (1997). "Spectrochemistry of solutions. part 29. Intrusion ion pairing: identification of a new form of ion pair in transition metal salt solutions in pyridine through their visible spectra". J. Mol. Liquids. 73-74: 99–106. doi:10.1016/S0167-7322(97)00060-3.
- ↑ Hans Falkenhagen, Theorie der Elektrolyte, S. Hirzel Verlag, Leipzig, 1971
- ↑ Klotz, I.M. (1964). Chemical Thermodynamics. W.A. Benjamin. Chapter 10
- ↑ Assuming that both Na+ and Cl− have six water molecules in the primary solvation shell at ambient temperatures a 5 M solution (5 mol dm−1) will consist almost entirely of fully solvated ion-pairs.
- ↑ Manfred Eigen, Nobel lecture
- ↑ Clifford, A.A. "Changes of water properties with temperature".
- ↑ Gans, P.; Gill, J.B.; Longdon, P.J (1989). "Spectrochemistry of solutions. Part 21.—Inner- and outer-sphere complexes of lithium with thiocyanate in acetonitrile solutions". J. Chem. Soc. Faraday Trans. 1. 85 (7): 1835–1839. doi:10.1039/F19898501835.
- ↑ Gans, P; Gill, J.B.; Holden, K.M.L. (1994). "Spectrochemistry of solutions. Part 27.—Formation of [Mg(NCS)]+ in solutions of Mg(NCS)2 in methanol". J. Chem. Soc. Faraday Trans. 90 (16): 2351–2352. doi:10.1039/FT9949002351.
- 1 2 3 4 5 Gill, J.B. (1981). "Solute-solute interactions in liquid ammonia solutions: a vibrational spectroscopic view". Pure Appl. Chem. 53 (7): 1365–1381. doi:10.1351/pac198153071365.
- ↑ Goralski, P.; Chabanel, M. (1987). "Vibrational study of ionic association in aprotic solvents. 11. Formation and structure of mixed aggregates between lithium halides and lithium thiocyanate". Inorg. Chem. 26 (13): 2169–2171. doi:10.1021/ic00260a032.
- ↑ Bacelon, P.; Corset, J.; de Loze , C. (2004). "Triple ion formation in solutions of alkaline sulfocyanides". J.Solution Chem. 9 (2): 129–139. doi:10.1007/BF00644484. (sulfocyanides = thiocyanates)
- ↑ Winstein, S.; Clippinger, E.; Fainberg, A. H.; Heck, R.; RobinsonG. C. (1956). "Salt Effects and Ion Pairs in Solvolysis and Related Reactions. III.1 Common Ion Rate Depression and Exchange of Anions during Acetolysis". Journal of the American Chemical Society. 78 (2): 328–335. doi:10.1021/ja01583a022.
- ↑ Davies, C.W. (1962). Ion Association. London: Butterworths.
- ↑ Miyoshi, K. (1973). "Comparison of the Conductance Equations of Fuoss - Onsager, Fuoss-Hsia and Pitts with the Data of Bis(2,9-dimethyl-1,10-phenanthroline)Cu(I) Perchlorate". Bull. Chem. Soc. Japan. 46 (2): 426–430. doi:10.1246/bcsj.46.426.
Further reading
Hans Falkenhagen, Theorie der Elektrolyte, S. Hirzel Verlag, Leipzig, 1971, chp 12, 16