Spectral line shape
Spectral line shape describes the form of a feature, observed in spectroscopy, corresponding to an energy change in an atom, molecule or ion. Ideal line shapes include Lorentzian, Gaussian and Voigt functions, whose parameters are the line position, maximum height and half-width.[1] Actual line shapes are determined principally by Doppler, collision and proximity broadening. For each system the half-width of the shape function varies with temperature, pressure (or concentration) and phase. A knowledge of shape function is needed for spectroscopic curve fitting and deconvolution.
Origins
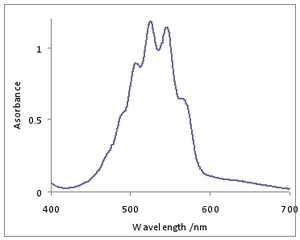
A spectroscopic transition is associated with a specific amount of energy, E. However, when this energy is measured by means of some spectroscopic technique, the spectroscopic line is not infinitely sharp, but has a particular shape. Numerous factors can contribute to the broadening of spectral lines. Broadening can only be mitigated by the use of specialized techniques, such as Lamb dip spectroscopy. The principal sources of broadening are:
- Lifetime broadening. According to the uncertainty principle the uncertainty in energy, ΔE and the lifetime, Δt, of the excited state are related by
- This determines the minimum possible line width. As the excited state decays exponentially in time this effect produces a line with Lorentzian shape in terms of frequency (or wavenumber).

- Doppler broadening. This is caused by the fact that the velocity of atoms or molecules relative to the observer follows a Maxwell distribution, so the effect is dependent on temperature. If this were the only effect the line shape would be Gaussian.[1]
- Pressure broadening (Collision broadening). Collisions between atoms or molecules reduce the lifetime of the upper state, Δt, increasing the uncertainty ΔE. This effect depends on both the density (that is, pressure for a gas) and the temperature, which affects the rate of collisions. The broadening effect is described by a Lorentzian profile in most cases.[2]
- Proximity broadening. The presence of other molecules close to the molecule involved affects both line width and line position. It is the dominant process for liquids and solids. An extreme example of this effect is the influence of hydrogen bonding on the spectra of protic liquids.
Observed spectral line shape and line width are also affected by instrumental factors. The observed line shape is a convolution of the intrinsic line shape with the instrument transfer function.[3]
Each of these mechanisms, and others, can act in isolation or in combination. If each effect is independent of the other, the observed line profile is a convolution of the line profiles of each mechanism. Thus, a combination of Doppler and pressure broadening effects yields a Voigt profile.
Line shape functions
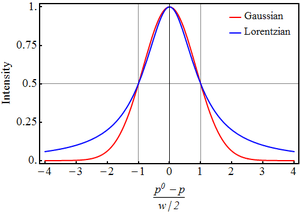
A Lorentzian line shape function can be represented as

where L signifies a Lorentzian function standardized, for spectroscopic purposes, to a maximum value of 1;[note 1] is a subsidiary variable defined as


where p0 is the position of the maximum (corresponding to the transition energy E), p is a position, and w is the full width at half maximum (FWHM), the width of the curve when the intensity is half the maximum intensity (this occurs at the points p = p0±w/2). The unit of p0, p and w is typically wavenumber or frequency. The variable x is dimensionless and is zero at p=p0.
The Gaussian line shape has the standardized form,

The subsidiary variable, x, is defined in the same way as for a Lorentzian shape. Both this function and the Lorentzian have a maximum value of 1 at x = 0 and a value of 1/2 at x=±1.
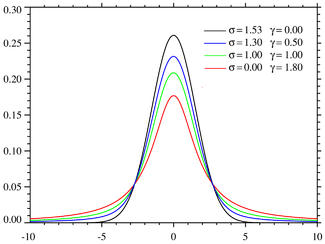
The third line shape that has a theoretical basis is the Voigt function, a convolution of a Gaussian and a Lorentzian,

where σ and γ are half-widths. The computation of a Voigt function and its derivatives is more complicated than a Gaussian or Lorentzian.[4]
A spectroscopic peak may be fitted to multiples of the above functions or to sums or products of functions with variable parameters.[5] The above functions are all symmetrical about the position of their maximum.[note 2] Asymmetric functions have also been used.[6][note 3]
Instances
Atomic spectra
For atoms in the gas phase the principal effects are Doppler and pressure broadening. Lines are relatively sharp on the scale of measurement so that applications such as atomic absorption spectroscopy (AAS) and Inductively coupled plasma atomic emission spectroscopy (ICP) are used for elemental analysis. Atoms also have distinct x-ray spectra that are attributable to the excitation of inner shell electrons to excited states. The lines are relatively sharp because the inner electron energies are not very sensitive to the atom's environment. This is applied to X-ray fluorescence spectroscopy of solid materials.
Molecular spectra
For molecules in the gas phase, the principal effects are Doppler and pressure broadening. This applies to rotational spectroscopy,[7] rotational-vibrational spectroscopy and vibronic spectroscopy.
For molecules in the liquid state or in solution, collision and proximity broadening predominate and lines are much broader than lines from the same molecule in the gas phase.[8][9] Line maxima may also be shifted. Because there are many sources of broadening, the lines have a stable distribution, tending towards a Gaussian shape.
Nuclear magnetic resonance
The shape of lines in a nuclear magnetic resonance (NMR) spectrum is determined by the process of free induction decay. This decay is approximately exponential, so the line shape is Lorentzian.[10] This follows because the Fourier transform of an exponential function in the time domain is a Lorentzian in the frequency domain. In NMR spectroscopy the lifetime of the excited states is relatively long, so the lines are very sharp, producing high-resolution spectra.
Magnetic resonance imaging

Gadolinium-based pharmaceuticals alter the relaxation time, and hence spectral line shape, of those protons that are in water molecules that are transiently attached to the paramagnetic atoms, resulting contrast enhancement of the MRI image.[11] This allows better visualisation of some brain tumours.[11]
Applications
Curve decomposition
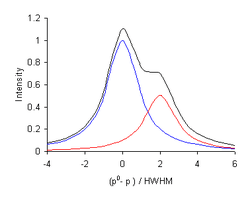
Some spectroscopic curves can be approximated by the sum of a set of component curves. For example, when Beer's law

applies, the measured intensity, I, at wavelength λ, is a linear combination of the intensity due to the individual components, k, at concentration, ck. ε is an extinction coefficient. In such cases the curve of experimental data may be decomposed into sum of component curves in a process of curve fitting. This process is also widely called deconvolution. Curve deconvolution and curve fitting are a completely different mathematical procedures.[6][12]
Curve fitting can be used in two distinct ways.
- The line shapes and parameters p0 and HWHM of the individual component curves have been obtained experimentally. In this case the curve may be decomposed using a linear least squares process simply to determine the concentrations of the components. This process is used in analytical chemistry to determine the composition of a mixture of the components of known molar absorptivity spectra. For example, if the heights of two lines are found to be h1 and h2, c1 = h1 / ε1 and c2 = h2 / ε2.[13]
- Parameters of the line shape are unknown. The intensity of each component is a function of at least 3 parameters, position, height and half-width. In addition one or both of the line shape function and baseline function may not be known with certainty. When two or more parameters of a fitting curve are not known the method of non-linear least squares must be used.[14][15] The reliability of curve fitting in this case is dependent on the separation between the components, their shape functions and relative heights, and the signal-to-noise ratio in the data.[6][16]
Derivative spectroscopy
Spectroscopic curves can be subjected to numerical differentiation.[17]
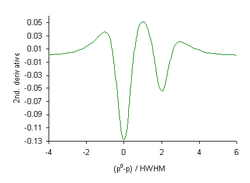
When the data points in a curve are equidistant from each other the Savitzky–Golay convolution method may be used.[18] The best convolution function to use depends primarily on the signal-to-noise-ratio of the data.[19] The first derivative (slope, ) of all single line shapes is zero at the position of maximum height. This is also true of the third derivative; odd derivatives can be used to locate the position of a peak maximum.[20]

The second derivatives, , of both Gaussian and Lorentzian functions have a reduced half-width. This can be used to apparently improve spectral resolution. The diagram shows the second derivative of the black curve in the diagram above it. Whereas the smaller component produces a shoulder in the spectrum, it appears as a separate peak in the 2nd. derivative.[note 4] Fourth derivatives, , can also be used, when the signal-to-noise-ratio in the spectrum is sufficiently high.[21]


Deconvolution
Deconvolution can be used to apparently improve spectral resolution. In the case of NMR spectra, the process is relatively straight forward, because the line shapes are Lorentzian, and the convolution of a Lorentzian with another Lorentzian is also Lorentzian. The Fourier transform of a Lorentzian is an exponential. In the co-domain (time) of the spectroscopic domain (frequency) convolution becomes multiplication. Therefore, a convolution of the sum of two Lorentzians becomes a multiplication of two exponentials in the co-domain. Since, in FT-NMR, the measurements are made in the time domain division of the data by an exponential is equivalent to deconvolution in the frequency domain. A suitable choice of exponential results in a reduction of the half-width of a line in the frequency domain. This technique has been rendered all but obsolete by advances in NMR technology.[22] A similar process has been applied for resolution enhancement of other types of spectra, with the disadvantage that the spectrum must be first Fourier transformed and then transformed back after the deconvoluting function has been applied in the spectrum's co-domain.[12]
See also
Notes
- ↑ In statistics Lorentzian (Cauchy) and Gaussian functions are normalized to unit area
- ↑ Experimental profiles that are symmetrical when plotted on a scale proportional to energy (for example, frequency or wavenumber) will not be symmetrical when plotted on a wavelength scale.
- ↑ In Electron paramagnetic resonance, asymmetric lines are characterized by two half-widths, measured either side of the line centre.
- ↑ Component peak maxima in the spectrum are minima in the 2nd. derivative spectrum and maxima in the 4th. derivative spectrum
References
- 1 2 Hollas, M.J. (1996). Modern Spectroscopy (3rd ed.). Wiley. pp. 30–34. ISBN 0471965227.
- ↑ Peach, g. (1981). "Theory of the pressure broadening and shift of spectral lines". Advances in Physics. 30 (3): 367–474. Bibcode:1981AdPhy..30..367P. doi:10.1080/00018738100101467.
- ↑ Gans, Section 9.3, Convolution and Cross-correlation
- ↑ Olivero, J.J.; R.L. Longbothum. "Empirical fits to the Voigt line width: A brief review". Journal of Quantitative Spectroscopy and Radiative Transfer. 17 (2): 233–236. Bibcode:1977JQSRT..17..233O. doi:10.1016/0022-4073(77)90161-3.
- ↑ Pitha, J.; Jones,R.N. (1966). "A Comparison of Optimization Methods for Fitting Curves to Infrared Band Envelopes". Canad. J. Chem. 44 (24): 3031–3050. doi:10.1139/v66-445.
- 1 2 3 Maddams, W.F. (1980). "The Scope and Limitations of Curve Fitting". Applied Spectroscopy. 34 (3): 245–267. Bibcode:1980ApSpe..34..245M. doi:10.1366/0003702804730312.
- ↑ Kroto, H.W. (1992). Molecular Rotation Spectra. Wiley. ISBN 0-486-49540-X. Section 4.6, Line shapes and line widths
- ↑ Clarke, J.H.R, "Band Shapes and Molecular Dynamics in liquids", pp. 109-193, in Advances in Infrared and Raman Spectroscopy, Volume 4 (1978), Editors Clark, R.J.H; Hester, R.E.
- ↑ Bradley, Michael S.; Bratu, Cheryl (1997). "Vibrational Line Profiles as a Probe of Molecular Interactions". J. Chem. Educ. 74 (5): 553. Bibcode:1997JChEd..74..553B. doi:10.1021/ed074p553.
- ↑ Petrakis, Leonidas (1967). "Spectral line shapes: Gaussian and Lorentzian functions in magnetic resonance". J. Chem. Educ. 44 (8): 432. Bibcode:1967JChEd..44..432P. doi:10.1021/ed044p432.
- 1 2 Brown, Mark A.; Semelka, Richard C. (2010). MRI: Basic Principles and Applications (4th ed.). Wiley-Blackwell. ISBN 978-0470500989. Chapter 3, Relaxation
- 1 2 Blass, W.E.; Halsey, G.W. (1981). Deconvolution of Absorption Spectra. Academic Press. ISBN 0121046508.
- ↑ Skoog, D.A.; West, D.H.; Holler, F.J.; Crouch, S.R. (2004). Fundamentals of Analytical Chemistry. Brooks/Cole. p. 796. ISBN 0-03-035523-0.
- ↑ Gans, Section 8.3, Gaussian, Lorentzian and related functions
- ↑ Sundius, T (1973). "Computer fitting of Voigt profiles to Raman lines". J. Raman Spectrosc. 1 (5): 457–488. Bibcode:1973JRSp....1..471S. doi:10.1002/jrs.1250010506.
- ↑ Gans, P; Gill, J.B. (1980). "Comments on the critical evaluation of curve fitting in infrared spectrometry". Anal. Chem. 52 (2): 351–352. doi:10.1021/ac50052a035.
- ↑ Bridge, T P; Fell. A.F; Wardman, R.H. (1987). "Perspectives in derivative spectroscopy Part 1-Theoretical principles". Journal of the Society of Dyers and Colourists. 103 (1): 17–27. doi:10.1111/j.1478-4408.1987.tb01081.x.
- ↑ Savitzky, A.; Golay, M.J.E. (1964). "Smoothing and Differentiation of data by Simplified Least Squares Procedures". Analytical Chemistry. 36 (8): 1627–1639. doi:10.1021/ac60214a047.
- ↑ Rzhevskii, Alexander M.; Mardilovich, Peter P. (1994). "Generalized GansGill Method for Smoothing and Differentiation of Composite Profiles in Practice". Applied Spectroscopy. 48 (1): 13–20. Bibcode:1994ApSpe..48...13R. doi:10.1366/0003702944027714.
- ↑ Gans, p. 158
- ↑ Antonov, Liudmil (1997). "Fourth derivative spectroscopy — a critical view". Analytica Chimica Acta. 349 (1-3): 295–301. doi:10.1016/S0003-2670(97)00210-9.
- ↑ Banwell, Colin N.; McCash, Elaine M. (1994). Fundamentals of molecular spectroscopy (4th ed.). McGraw-Hill. p. 40. ISBN 0-07-707976-0.Section 7.2.6, Simplification of Complex Spectra.
Further reading
- Atkins, P.W.; de Paula, J. (2006). "13.3: Linewidths". Physical Chemistry (8th ed.). Oxford University Press. pp. 436–438. ISBN 0198700725.
- Davies, Christopher C. (2000). Lasers and electro-optics : fundamentals and engineering (Reprint ed.). New York: Cambridge University Press. pp. 27–30. ISBN 9780521484039.
- Demtröder, Wolfgang (2008). Laser spectroscopy. basic principles (4th ed.). Drodrecht: Springer-Verlag. pp. 88–89. ISBN 9783540734185.
- De Natale, Paolo; Pasquale Maddaloni; Marco Bellini (2013). Laser-based Measurements for Time and Frequency Domain Applications. Taylor & Francis. pp. 181–183. ISBN 9781439841518.
- Gans, P. (1992). Data Fitting in the Chemical Sciences. Wiley. ISBN 0 471 93412 7.
- Hewitt, John (May 13, 2013). "Manipulating Lorentz and Fano spectral line shapes". phys.org. Retrieved May 25, 2013.
- Linne, Mark A. (2002). "11. Line broadening". Spectroscopic measurement an introduction to the fundamentals. Amsterdam: Academic Press. pp. 283–303. ISBN 9780080517537.
- Pelikan, Peter; Ceppan, Michal; Liska, Marek (1994). Applications of Numerical Methods in Molecular Spectroscopy. CRC Press. ISBN 9780849373220.
- Poole, Jr., Charles P. (2004). "Line shape". Encyclopedic Dictionary of Condensed Matter Physics. 1. Academic Press. pp. 718–719. ISBN 9780080545233.
- Simmons, Joseph H.; Potter, Kelly S. (2000). Optical materials ([Online-Ausg.] ed.). San Diego, Calif.: Academic. pp. 274–277. ISBN 9780126441406.
- Telle, Helmut H; Angel González Ureña; Robert J. Donovan (2007). Laser chemistry spectroscopy, dynamics and applications. Chichester, West Sussex, England: John Wiley & Sons. pp. 24–25. ISBN 9780470059401.
External links
- Curve Fitting in Raman and IR Spectroscopy: Basic Theory of Line Shapes and Applications
- 21st International Conference on Spectral Line Shapes, St. Petersburg (2012)