Relaxation (NMR)
In nuclear magnetic resonance (NMR) spectroscopy and magnetic resonance imaging (MRI) the term relaxation describes how signals change with time. In general signals deteriorate with time, becoming weaker and broader. The deterioration reflects the fact that the NMR signal, which results from nuclear magnetization, arises from the over-population of an excited state. Relaxation is the conversion of this non-equilibrium population to a normal population. In other words, relaxation describes how quickly spins "forget" the direction in which they are oriented. The rates of this spin relaxation can be measured in both spectroscopy and imaging applications.[1]
T1 and T2
The deterioration of an NMR signal is analyzed in terms of two separate processes, each with their own time constants. One process, associated with T1, is responsible for the loss of signal intensity. The other process, associated with T2, is responsible for the broadening of the signal. Stated more formally, T1 is the time constant for the physical processes responsible for the relaxation of the components of the nuclear spin magnetization vector M parallel to the external magnetic field, B0 (which is conventionally oriented along the z axis). T2 relaxation affects the components of M perpendicular to B0. In conventional NMR spectroscopy T1 determines the recycle time, the rate at which an NMR spectrum can be acquired. Values of T1 range from milliseconds to several seconds.
T1
The longitudinal (or spin-lattice) relaxation time T1 is the decay constant for the recovery of the z component of the nuclear spin magnetization, Mz, towards its thermal equilibrium value, . In general,

![{\displaystyle M_{z}(t)=M_{z,\mathrm {eq} }(0)-[M_{z,\mathrm {eq} }-M_{z}(0)]e^{-t/T_{1}}}](../I/m/60539fa5aa90acc12c7f1c8c8103693fbbb237b0.svg)
In specific cases:
- If M has been tilted into the xy plane, then and the recovery is simply


i.e. the magnetization recovers to 63% of its equilibrium value after one time constant T1.
- In the inversion recovery experiment, commonly used to measure T1 values, the initial magnetization is inverted, , and so the recovery follows


T1 relaxation involves redistributing the populations of the nuclear spin states in order to reach the thermal equilibrium distribution. By definition, this is not energy conserving. Moreover, spontaneous emission is negligibly slow at NMR frequencies. Hence truly isolated nuclear spins would show negligible rates of T1 relaxation. However, a variety of relaxation mechanisms allow nuclear spins to exchange energy with their surroundings, the lattice, allowing the spin populations to equilibrate. The fact that T1 relaxation involves an interaction with the surroundings is the origin of the alternative description, spin-lattice relaxation.
Note that the rates of T1 relaxation (i.e., 1/T1) are generally strongly dependent on the NMR frequency and so vary considerably with magnetic field strength B. Small amounts of paramagnetic substances in a sample speed up relaxation very much. By degassing, and thereby removing dissolved Oxygen, the T1/T2 of liquid samples easily go up to an order of ten seconds.
Spin saturation transfer
Especially for molecules exhibiting slowly relaxing (T1) signals, the technique spin saturation transfer (SST) provides information on chemical exchange reactions. The method is widely applicable to fluxional molecules. This magnetization transfer technique provides rates, provided that they exceed 1/T1.[2]
T2
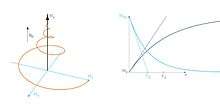


The transverse (or spin-spin) relaxation time T2 is the decay constant for the component of M perpendicular to B0, designated Mxy, MT, or . For instance, initial xy magnetization at time zero will decay to zero (i.e. equilibrium) as follows:


i.e. the transverse magnetization vector drops to 37% of its original magnitude after one time constant T2.
T2 relaxation is a complex phenomenon, but at its most fundamental level, it corresponds to a decoherence of the transverse nuclear spin magnetization. Random fluctuations of the local magnetic field lead to random variations in the instantaneous NMR precession frequency of different spins. As a result, the initial phase coherence of the nuclear spins is lost, until eventually the phases are disordered and there is no net xy magnetization. Because T2 relaxation involves only the phases of other nuclear spins it is often called "spin-spin" relaxation.
T2 values are generally much less dependent on field strength, B, than T1 values.
A Hahn echo decay experiment can be used to measure the T2 time, as shown in the animation below. The size of the echo is recorded for different spacings of the two applied pulses. This reveals the decoherence which is not refocused by the 180° pulse. In simple cases, an exponential decay is measured which is described by the time.
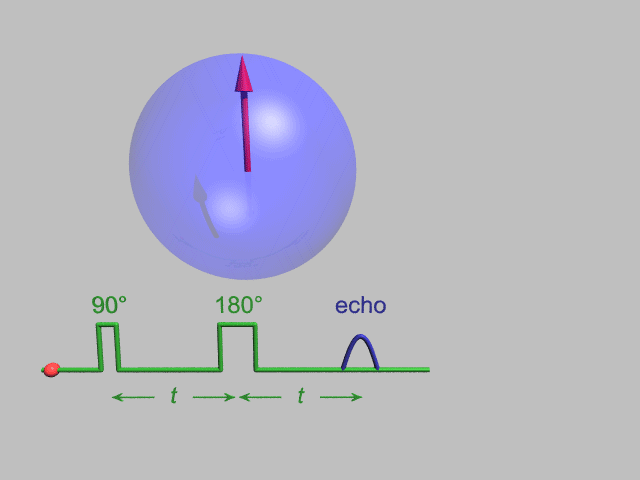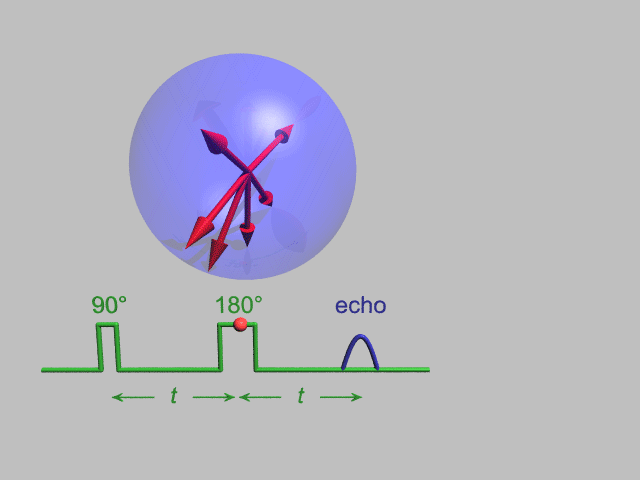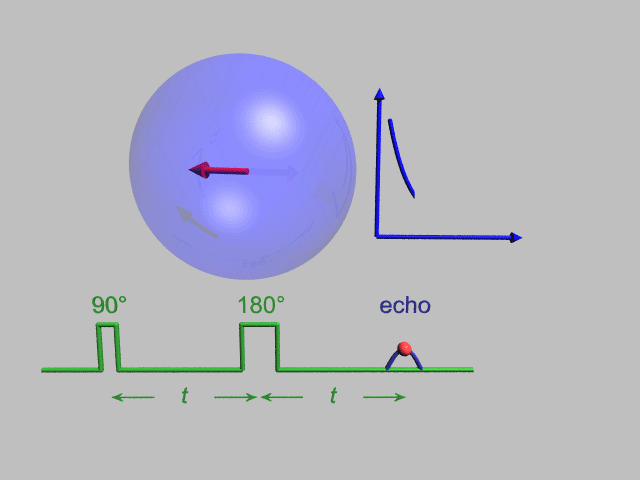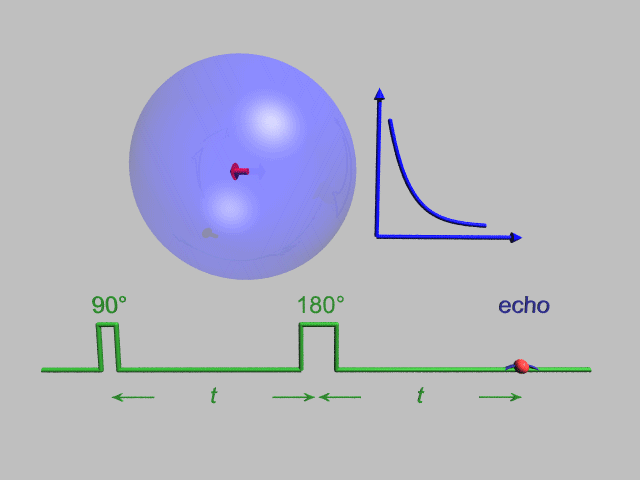
T2* and magnetic field inhomogeneity
In an idealized system, all nuclei in a given chemical environment, in a magnetic field, precess with the same frequency. However, in real systems, there are minor differences in chemical environment which can lead to a distribution of resonance frequencies around the ideal. Over time, this distribution can lead to a dispersion of the tight distribution of magnetic spin vectors, and loss of signal (Free Induction Decay). In fact, for most magnetic resonance experiments, this "relaxation" dominates. This results in dephasing.
However, decoherence because of magnetic field inhomogeneity is not a true "relaxation" process; it is not random, but dependent on the location of the molecule in the magnet. For molecules that aren't moving, the deviation from ideal relaxation is consistent over time, and the signal can be recovered by performing a spin echo experiment.
The corresponding transverse relaxation time constant is thus T2*, which is usually much smaller than T2. The relation between them is:

where γ represents gyromagnetic ratio, and ΔB0 the difference in strength of the locally varying field.
Unlike T2, T2* is influenced by magnetic field gradient irregularities. The T2* relaxation time is always shorter than the T2 relaxation time and is typically milliseconds for water samples in imaging magnets.
Is T1 always longer than T2?
The following always holds true:[3] . In most situations (but not in principle) is greater than .

Bloch equations
Bloch equations are used to calculate the nuclear magnetization M = (Mx, My, Mz) as a function of time when relaxation times T1 and T2 are present. Bloch equations are phenomenological equations that were introduced by Felix Bloch in 1946.[4]



Where γ is the gyromagnetic ratio and B(t) = (Bx(t), By(t), B0 + Bz(t)) is the magnetic flux density experienced by the nuclei. The z component of the magnetic flux density B is typically composed of two terms: one, B0, is constant in time, the other one, Bz(t), is time dependent. It is present in magnetic resonance imaging and helps with the spatial decoding of the NMR signal. M(t) × B(t) is the cross product of these two vectors.
The equation listed above in the section on T1 and T2 relaxation can be derived from Bloch equations.
Solomon equations
Solomon equations are used to calculate the transfer of magnetization as a result of relaxation in a dipolar system. They can be employed to explain the nuclear Overhauser effect, which is an important tool in determining molecular structure.
Common relaxation time constants in human tissues
Following is a table of the approximate values of the two relaxation time constants for hydrogen nuclear spins in nonpathological human tissues.
| Tissue type | Approximate T1 value in ms | Approximate T2 value in ms |
|---|---|---|
| Adipose tissues | 240-250 | 60-80 |
| Whole blood (deoxygenated) | 1350 | 50 |
| Whole blood (oxygenated) | 1350 | 200 |
| Cerebrospinal fluid (similar to pure water) | 4200 - 4500 | 2100-2300 |
| Gray matter of cerebrum | 920 | 100 |
| White matter of cerebrum | 780 | 90 |
| Liver | 490 | 40 |
| Kidneys | 650 | 60-75 |
| Muscles | 860-900 | 50 |
Following is a table of the approximate values of the two relaxation time constants for chemicals that commonly show up in human brain magnetic resonance spectroscopy (MRS) studies, physiologically or pathologically.
| Signals of chemical groups | Relative resonance frequency | Approximate T1 value (ms) | Approximate T2 value (ms) |
|---|---|---|---|
| Creatine (Cr) and Phosphocreatine (PCr)[5] | 3.0 ppm | gray matter: 1150-1340, white matter: 1050-1360 |
gray matter: 198-207, white matter: 194-218 |
| N-Acetyl group (NA), mainly from N-Acetylaspartate (NAA)[5] |
2.0 ppm | gray matter: 1170-1370, white matter: 1220-1410 |
gray matter: 388-426, white matter: 436-519 |
| —CH3 group of Lactate[6] |
1.33 ppm (doublet: 1.27 & 1.39 ppm) |
(To be listed) | 1040 |
Relaxation in the rotating frame, T1ρ
The discussion above describes relaxation of nuclear magnetization in the presence of a constant magnetic field B0. This is called relaxation in the laboratory frame. Another technique, called relaxation in the rotating frame, is the relaxation of nuclear magnetization in the presence of the field B0 together with a time-dependent magnetic field B1. The field B1 rotates in the plane perpendicular to B0 at the Larmor frequency of the nuclei in the B0. The magnitude of B1 is typically much smaller than the magnitude of B0. Under these circumstances the relaxation of the magnetization is similar to laboratory frame relaxation in a field B1. The decay constant for the recovery of the magnetization component along B1 is called the spin-lattice relaxation time in the rotating frame and is denoted T1ρ. Relaxation in the rotating frame is useful because it provides information on slow motions of nuclei.
Microscopic mechanisms
Relaxation of nuclear spins requires a microscopic mechanism for a nucleus to change orientation with respect to the applied magnetic field and/or interchange energy with the surroundings (called the lattice). The most common mechanism is the magnetic dipole-dipole interaction between the magnetic moment of a nucleus and the magnetic moment of another nucleus or other entity (electron, atom, ion, molecule). This interaction depends on the distance between the pair of dipoles (spins) but also on their orientation relative to the external magnetic field. Several other relaxation mechanisms also exist. The chemical shift anisotropy (CSA) relaxation mechanism arises whenever the electronic environment around the nucleus is non spherical, the magnitude of the electronic shielding of the nucleus will then be dependent on the molecular orientation relative to the (fixed) external magnetic field. The spin rotation (SR) relaxation mechanism arises from an interaction between the nuclear spin and a coupling to the overall molecular rotational angular momentum. Nuclei with spin I ≥ 1 will have not only a nuclear dipole but a quadrupole. The nuclear quadrupole has an interaction with the electric field gradient at the nucleus which is again orientation dependent as with the other mechanisms described above, leading to the so-called quadrupolar relaxation mechanism.
Molecular reorientation or tumbling can then modulate these orientation-dependent spin interaction energies. According to quantum mechanics, time-dependent interaction energies cause transitions between the nuclear spin states which result in nuclear spin relaxation. The application of time-dependent perturbation theory in quantum mechanics shows that the relaxation rates (and times) depend on spectral density functions that are the Fourier transforms of the autocorrelation function of the fluctuating magnetic dipole interactions.[7] The form of the spectral density functions depend on the physical system, but a simple approximation called the BPP theory is widely used.
Another relaxation mechanism is the electrostatic interaction between a nucleus with an electric quadrupole moment and the electric field gradient that exists at the nuclear site due to surrounding charges. Thermal motion of a nucleus can result in fluctuating electrostatic interaction energies. These fluctuations produce transitions between the nuclear spin states in a similar manner to the magnetic dipole-dipole interaction.
BPP theory
In 1948, Nicolaas Bloembergen, Edward Mills Purcell, and Robert Pound proposed the so-called Bloembergen-Purcell-Pound theory (BPP theory) to explain the relaxation constant of a pure substance in correspondence with its state, taking into account the effect of tumbling motion of molecules on the local magnetic field disturbance.[8] The theory was in good agreement with experiments on pure substances, but not for complicated environments such as the human body.
This theory makes the assumption that the autocorrelation function of the microscopic fluctuations causing the relaxation is proportional to , where is called the correlation time. From this theory, one can get T1 > T2 for magnetic dipolar relaxation:


- ,
![\frac{1}{T_1}=K[\frac{\tau_c}{1+\omega_0^2\tau_c^2}+\frac{4\tau_c}{1+4\omega_0^2\tau_c^2}]](../I/m/1e2dc41c30f68afa5be0c9835bbddfdbc0e07bfd.svg)
![\frac{1}{T_2}=\frac{K}{2}[3\tau_c+\frac{5\tau_c}{1+\omega_0^2\tau_c^2}+\frac{2\tau_c}{1+4\omega_0^2\tau_c^2}]](../I/m/8d66b2c145e6bdd0434e86a181a19b91fd879c08.svg)
where is the Larmor frequency in correspondence with the strength of the main magnetic field . is the correlation time of the molecular tumbling motion. is defined for spin-1/2 nuclei and is a constant with being the magnetic permeability of free space of the the reduced Planck constant, γ the gyromagnetic ratio of such species of nuclei, and r the distance between the two nuclei carrying magnetic dipole moment.





Taking for example the H2O molecules in liquid phase without the contamination of oxygen-17, the value of K is 1.02×1010 s−2 and the correlation time is on the order of picoseconds = s, while hydrogen nuclei 1H (protons) at 1.5 teslas carry an Larmor frequency of approximately 64 MHz. We can then estimate using τc = 5×10−12 s:

- (dimensionless)
- = 3.92 s
- = 3.92 s,

![T_1=\left(1.02\times 10^{10}\left[\frac{ 5\times 10^{-12} }{1 + (3.2\times 10^{-5} )^2} + \frac{ 4\cdot 5\times 10^{-12} }{1 + 4\cdot (3.2\times 10^{-5} )^2}\right]\right)^{-1}](../I/m/64afdc9753fa069531dd42c3446cc4f25013feed.svg)
![T_2=\left(\frac{1.02\times 10^{10}}{2}\left[3\cdot 5\times 10^{-12} + \frac{5\cdot 5\times 10^{-12} }{1 + \left(3.2\times 10^{-5} \right)^2} + \frac{ 2\cdot 5\times 10^{-12} }{1 + 4\cdot (3.2\times 10^{-5} )^2}\right]\right)^{-1}](../I/m/76347084bfc4dfd43a96f8e5eed5bfb421261ca2.svg)
which is close to the experimental value, 3.6 s. Meanwhile, we can see that at this extreme case, T1 equals T2. As follows from the BPP theory, measuring the T1 times leads to internuclear distances r. One of the examples is accurate determinations of the metal – hydride (M-H) bond lengths in solutions by measurements of 1H selective and non-selective T1 times in variable-temperature relaxation experiments via equation [9][10]
r(M-H) (Å ) = C((1.4k + 4.47) T1min / ν)1/6
k = (f-1)/(0.5-f/3) with f = T1s/T1
C = 107(γH2 γM2 ħ2 I(I +1) / 15 ) 1/6
I is spin of M
where frequency and T1 are measured in MHz and s, respectively.
See also
- Nuclear magnetic resonance
- Nuclear magnetic resonance spectroscopy of carbohydrates
- Nuclear magnetic resonance spectroscopy of nucleic acids
- Nuclear magnetic resonance spectroscopy of proteins
- NMR spectroscopy
- Protein dynamics
- Relaxometry
References
- ↑ Friebolin, H., "Basic One- and Two- Dimensional NMR Spectroscopy, 4th ed.," VCH: Weinheim, 2008. ISBN 978-3-527-31233-7
- ↑ Jarek, R. L., Flesher, R. J., Shin, S. K., "Kinetics of Internal Rotation of N,N-Dimethylacetamide: A Spin-Saturation Transfer Experiment", Journal of Chemical Education 1997, volume 74, page 978. doi:10.1021/ed074p978.
- ↑ Malcolm H. Levitt: Spin Dynamics: Basics of Nuclear Magnetic Resonance, 2nd edition, John Wiley & Sons, New York 2008, ISBN 0-470-51117-6, Section 11.9.2
- ↑ F Bloch, Nuclear Induction, Physical Review 70, 460-473 (1946)
- 1 2 Chemicals of brain relaxation time at 1.5T. Kreis R, Ernst T, and Ross BD "Absolute Quantification of Water and Metabolites in the Human Brain. II. Metabolite Concentrations" Journal of Magnetic Resonance, Series B 102 (1993): 9-19
- ↑ Lactate relaxation time at 1.5 T. Isobe T, Matsumura A, Anno I, Kawamura H, Muraishi H, Umeda T, Nose T. "Effect of J coupling and T2 Relaxation in Assessing of Methyl Lactate Signal using PRESS Sequence MR Spectroscopy." Igaku Butsuri (2005) v25. 2:68-74.
- ↑ A. Abragam "Principles of Nuclear Magnetism" (Oxford University Press, 1961)
- ↑ Bloembergen, E.M. Purcell, R.V. Pound "Relaxation Effects in Nuclear Magnetic Resonance Absorption" Physical Review (1948) v73. 7:679-746
- ↑ Dmitry G. Gusev, Daniel Nietlispach, Alexey B. Vymenits, Vladimir I. Bakhmutov, Heinz Berke Synthesis and NMR T1 relaxation study of rhenium and manganese hydride complexes
- ↑ D. G. Gusev, A. B. Vymenits, V. I. Bakhmutov Short spin-lattice relaxation times of hydride ligands. Proton-metal dipole-dipole interactions Inorg. Chem., 1991, 30 (16), p. 3116. DOI: 10.1021/ic00016a003Inorg. Chem., 1993, 32 (15), p. 3270. doi:10.1021/ic00067a013
External links
- basics of NMR
- Relaxation in high-resolution NMR spectroscopy
- Field-cycling NMR relaxometry
- relax Software for the analysis of NMR dynamics
- Estimation of T1 and T2 relaxation parameters in MRI