Cystic fibrosis
| Cystic fibrosis | |
|---|---|
| Synonyms | mucoviscidosis |
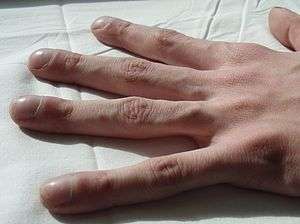 | |
| Clubbing in the fingers of a person with cystic fibrosis | |
| Classification and external resources | |
| Specialty | Medical genetics, pulmonology |
| ICD-10 | E84 |
| ICD-9-CM | 277.0 |
| OMIM | 219700 |
| DiseasesDB | 3347 |
| MedlinePlus | 000107 |
| eMedicine | article/1001602 |
| Patient UK | Cystic fibrosis |
| MeSH | D003550 |
| GeneReviews | |
| Orphanet | 586 |
Cystic fibrosis (CF) is a genetic disorder that affects mostly the lungs, but also the pancreas, liver, kidneys, and intestine.[1][2] Long-term issues include difficulty breathing and coughing up mucus as a result of frequent lung infections. Other signs and symptoms include sinus infections, poor growth, fatty stool, clubbing of the fingers and toes, and infertility in males, among others. Different people may have different degrees of symptoms.[1]
CF is inherited in an autosomal recessive manner. It is caused by the presence of mutations in both copies of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein.[1] Those with a single working copy are carriers and otherwise mostly normal.[3] CFTR is involved in production of sweat, digestive fluids, and mucus.[4] When CFTR is not functional, secretions which are usually thin instead become thick.[5] The condition is diagnosed by a sweat test and genetic testing.[1] Screening of infants at birth takes place in some areas of the world.[1]
No cure for cystic fibrosis is known.[3] Lung infections are treated with antibiotics which may be given intravenously, inhaled, or by mouth. Sometimes, the antibiotic azithromycin is used long term. Inhaled hypertonic saline and salbutamol may also be useful. Lung transplantation may be an option if lung function continues to worsen. Pancreatic enzyme replacement and fat-soluble vitamin supplementation are important, especially in the young.[1] Airway clearance techniques such as chest physiotherapy have some short-term benefit, but long-term effects are unclear.[6] The average life expectancy is between 42 and 50 years in the developed world.[7][8] Lung problems are responsible for death in 80% of people with cystic fibrosis.[1]
CF is most common among people of Northern European ancestry and affects about one out of every 3,000 newborns.[1] About one in 25 people is a carrier.[3] It is least common in Africans and Asians.[1] It was first recognized as a specific disease by Dorothy Andersen in 1938, with descriptions that fit the condition occurring at least as far back as 1595.[2] The name 'cystic fibrosis' refers to the characteristic fibrosis and cysts that form within the pancreas.[2][9]

Signs and symptoms
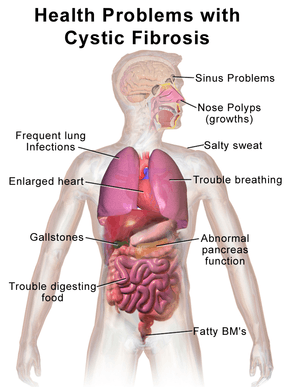
The main signs and symptoms of cystic fibrosis are salty-tasting skin,[10] poor growth, and poor weight gain despite normal food intake,[11] accumulation of thick, sticky mucus,[12] frequent chest infections, and coughing or shortness of breath.[13] Males can be infertile due to congenital absence of the vas deferens.[14] Symptoms often appear in infancy and childhood, such as bowel obstruction due to meconium ileus in newborn babies.[15] As the children grow, they exercise to release mucus in the alveoli.[16] Ciliated epithelial cells in the person have a mutated protein that leads to abnormally viscous mucus production.[12] The poor growth in children typically presents as an inability to gain weight or height at the same rate as their peers, and is occasionally not diagnosed until investigation is initiated for poor growth. The causes of growth failure are multifactorial and include chronic lung infection, poor absorption of nutrients through the gastrointestinal tract, and increased metabolic demand due to chronic illness.[11]
In rare cases, cystic fibrosis can manifest itself as a coagulation disorder. Vitamin K is normally absorbed from breast milk, formula, and later, solid foods. This absorption is impaired in some cystic fibrosis patients. Young children are especially sensitive to vitamin K malabsorptive disorders because only a very small amount of vitamin K crosses the placenta, leaving the child with very low reserves and limited ability to absorb vitamin K from dietary sources after birth. Because factors II, VII, IX, and X (clotting factors) are vitamin K–dependent, low levels of vitamin K can result in coagulation problems. Consequently, when a child presents with unexplained bruising, a coagulation evaluation may be warranted to determine whether an underlying disease is present.[17]
Lungs and sinuses
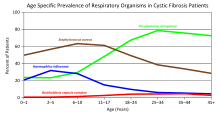
Green = Pseudomonas aeruginosa
Brown = Staphylococcus aureus
Blue = Haemophilus influenzae
Red = Burkholderia cepacia complex
Lung disease results from clogging of the airways due to mucus build-up, decreased mucociliary clearance, and resulting inflammation.[18][19] Inflammation and infection cause injury and structural changes to the lungs, leading to a variety of symptoms. In the early stages, incessant coughing, copious phlegm production, and decreased ability to exercise are common. Many of these symptoms occur when bacteria that normally inhabit the thick mucus grow out of control and cause pneumonia. In later stages, changes in the architecture of the lung, such as pathology in the major airways (bronchiectasis), further exacerbate difficulties in breathing. Other signs include coughing up blood (hemoptysis), high blood pressure in the lung (pulmonary hypertension), heart failure, difficulties getting enough oxygen to the body (hypoxia), and respiratory failure requiring support with breathing masks, such as bilevel positive airway pressure machines or ventilators.[20] Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa are the three most common organisms causing lung infections in CF patients.[19] In addition to typical bacterial infections, people with CF more commonly develop other types of lung disease. Among these is allergic bronchopulmonary aspergillosis, in which the body's response to the common fungus Aspergillus fumigatus causes worsening of breathing problems. Another is infection with Mycobacterium avium complex, a group of bacteria related to tuberculosis, which can cause lung damage and does not respond to common antibiotics.[21]
Mucus in the paranasal sinuses is equally thick and may also cause blockage of the sinus passages, leading to infection. This may cause facial pain, fever, nasal drainage, and headaches. Individuals with CF may develop overgrowth of the nasal tissue (nasal polyps) due to inflammation from chronic sinus infections.[22] Recurrent sinonasal polyps can occur in 10% to 25% of CF patients.[19] These polyps can block the nasal passages and increase breathing difficulties.[23][24]
Cardiorespiratory complications are the most common cause of death (about 80%) in patients at most CF centers in the United States.[19]
Gastrointestinal
Prior to prenatal and newborn screening, cystic fibrosis was often diagnosed when a newborn infant failed to pass feces (meconium). Meconium may completely block the intestines and cause serious illness. This condition, called meconium ileus, occurs in 5–10%[19][25] of newborns with CF. In addition, protrusion of internal rectal membranes (rectal prolapse) is more common, occurring in as many as 10% of children with CF,[19] and it is caused by increased fecal volume, malnutrition, and increased intra–abdominal pressure due to coughing.[26]
The thick mucus seen in the lungs has a counterpart in thickened secretions from the pancreas, an organ responsible for providing digestive juices that help break down food. These secretions block the exocrine movement of the digestive enzymes into the duodenum and result in irreversible damage to the pancreas, often with painful inflammation (pancreatitis).[27] The pancreatic ducts are totally plugged in more advanced cases, usually seen in older children or adolescents.[19] This causes atrophy of the exocrine glands and progressive fibrosis.[19]
The lack of digestive enzymes leads to difficulty absorbing nutrients with their subsequent excretion in the feces, a disorder known as malabsorption. Malabsorption leads to malnutrition and poor growth and development because of calorie loss. Resultant hypoproteinemia may be severe enough to cause generalized edema.[19] Individuals with CF also have difficulties absorbing the fat-soluble vitamins A, D, E, and K.
In addition to the pancreas problems, people with cystic fibrosis experience more heartburn, intestinal blockage by intussusception, and constipation.[28] Older individuals with CF may develop distal intestinal obstruction syndrome when thickened feces cause intestinal blockage.[29]
Exocrine pancreatic insufficiency occurs in the majority (85% to 90%) of patients with CF.[19] It is mainly associated with "severe" CFTR mutations, where both alleles are completely nonfunctional (e.g. ΔF508/ΔF508).[19] It occurs in 10% to 15% of patients with one "severe" and one "mild" CFTR mutation where little CFTR activity still occurs, or where two "mild" CFTR mutations exist.[19] In these milder cases, sufficient pancreatic exocrine function is still present so that enzyme supplementation is not required.[19] Usually, no other GI complications occur in pancreas-sufficient phenotypes, and in general, such individuals usually have excellent growth and development.[19] Despite this, idiopathic chronic pancreatitis can occur in a subset of pancreas-sufficient individuals with CF, and is associated with recurrent abdominal pain and life-threatening complications.[19]
Thickened secretions also may cause liver problems in patients with CF. Bile secreted by the liver to aid in digestion may block the bile ducts, leading to liver damage. Over time, this can lead to scarring and nodularity (cirrhosis). The liver fails to rid the blood of toxins and does not make important proteins, such as those responsible for blood clotting.[30][31] Liver disease is the third-most common cause of death associated with CF.[19]
Endocrine
The pancreas contains the islets of Langerhans, which are responsible for making insulin, a hormone that helps regulate blood glucose. Damage of the pancreas can lead to loss of the islet cells, leading to a type of diabetes unique to those with the disease.[32] This cystic fibrosis-related diabetes shares characteristics that can be found in type 1 and type 2 diabetics, and is one of the principal nonpulmonary complications of CF.[33] Vitamin D is involved in calcium and phosphate regulation. Poor uptake of vitamin D from the diet because of malabsorption can lead to the bone disease osteoporosis in which weakened bones are more susceptible to fractures.[34] In addition, people with CF often develop clubbing of their fingers and toes due to the effects of chronic illness and low oxygen in their tissues.[35][36]
Infertility
Infertility affects both men and women. At least 97% of men with cystic fibrosis are infertile, but not sterile and can have children with assisted reproductive techniques.[37] The main cause of infertility in men with CF is congenital absence of the vas deferens (which normally connects the testes to the ejaculatory ducts of the penis), but potentially also by other mechanisms such as causing no sperm, teratospermia, and few sperm with poor motility.[38] Many men found to have congenital absence of the vas deferens during evaluation for infertility have a mild, previously undiagnosed form of CF.[39] Around 20% of women with CF have fertility difficulties due to thickened cervical mucus or malnutrition. In severe cases, malnutrition disrupts ovulation and causes a lack of menstruation.[40]
Cause

CF is caused by a mutation in the gene cystic fibrosis transmembrane conductance regulator (CFTR). The most common mutation, ΔF508, is a deletion (Δ signifying deletion) of three nucleotides[41] that results in a loss of the amino acid phenylalanine (F) at the 508th position on the protein. This mutation accounts for two-thirds (66–70%[19]) of CF cases worldwide and 90% of cases in the United States; however, over 1500 other mutations can produce CF.[42] Although most people have two working copies (alleles) of the CFTR gene, only one is needed to prevent cystic fibrosis. CF develops when neither allele can produce a functional CFTR protein. Thus, CF is considered an autosomal recessive disease.
The CFTR gene, found at the q31.2 locus of chromosome 7, is 230,000 base pairs long, and creates a protein that is 1,480 amino acids long. More specifically, the location is between base pair 117,120,016 and 117,308,718 on the long arm of chromosome 7, region 3, band 1, subband 2, represented as 7q31.2. Structurally, CFTR is a type of gene known as an ABC gene. The product of this gene (the CFTR protein) is a chloride ion channel important in creating sweat, digestive juices, and mucus. This protein possesses two ATP-hydrolyzing domains, which allows the protein to use energy in the form of ATP. It also contains two domains comprising six alpha helices apiece, which allow the protein to cross the cell membrane. A regulatory binding site on the protein allows activation by phosphorylation, mainly by cAMP-dependent protein kinase.[20] The carboxyl terminal of the protein is anchored to the cytoskeleton by a PDZ domain interaction.[43]
In addition, the evidence is increasing that genetic modifiers besides CFTR modulate the frequency and severity of the disease. One example is mannan-binding lectin, which is involved in innate immunity by facilitating phagocytosis of microorganisms. Polymorphisms in one or both mannan-binding lectin alleles that result in lower circulating levels of the protein are associated with a threefold higher risk of end-stage lung disease, as well as an increased burden of chronic bacterial infections.[19]
Pathophysiology
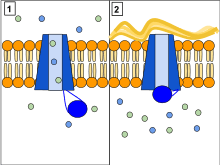
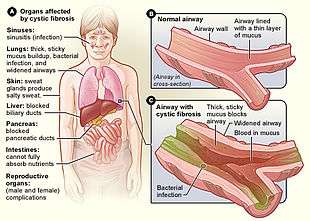
Several mutations in the CFTR gene can occur, and different mutations cause different defects in the CFTR protein, sometimes causing a milder or more severe disease. These protein defects are also targets for drugs which can sometimes restore their function. ΔF508-CFTR, which occurs in >90% of patients in the U.S., creates a protein that does not fold normally and is not appropriately transported to the cell membrane, resulting in its degradation. Other mutations result in proteins that are too short (truncated) because production is ended prematurely. Other mutations produce proteins that do not use energy normally, do not allow chloride, iodide, and thiocyanate to cross the membrane appropriately,[44] and degrade at a faster rate than normal. Mutations may also lead to fewer copies of the CFTR protein being produced.[20]
The protein created by this gene is anchored to the outer membrane of cells in the sweat glands, lungs, pancreas, and all other remaining exocrine glands in the body. The protein spans this membrane and acts as a channel connecting the inner part of the cell (cytoplasm) to the surrounding fluid. This channel is primarily responsible for controlling the movement of halogens from inside to outside of the cell; however, in the sweat ducts, it facilitates the movement of chloride from the sweat duct into the cytoplasm. When the CFTR protein does not resorb ions in sweat ducts, chloride and thiocyanate[45] released from sweat glands are trapped inside the ducts and pumped to the skin. Additionally hypothiocyanite, OSCN, cannot be produced by the immune defense system.[46][47] Because chloride is negatively charged, this modifies the electrical potential inside and outside the cell that normally causes cations to cross into the cell. Sodium is the most common cation in the extracellular space. The excess chloride within sweat ducts prevents sodium resorption by epithelial sodium channels and the combination of sodium and chloride creates the salt, which is lost in high amounts in the sweat of individuals with CF. This lost salt forms the basis for the sweat test.[20]
Most of the damage in CF is due to blockage of the narrow passages of affected organs with thickened secretions. These blockages lead to remodeling and infection in the lung, damage by accumulated digestive enzymes in the pancreas, blockage of the intestines by thick feces, etc. Several theories have been posited on how the defects in the protein and cellular function cause the clinical effects. The most current theory suggests that defective ion transport leads to dehydration in the airway epithelia, thickening mucus. In airway epithelial cells, the cilia exist in between the cell's apical surface and mucus in a layer known as airway surface liquid (ASL). The flow of ions from the cell and into this layer is determined by ion channels such as CFTR. CFTR not only allows chloride ions to be drawn from the cell and into the ASL, but it also regulates another channel called ENac, which allows sodium ions to leave the ASL and enter the respiratory epithelium. CFTR normally inhibits this channel, but if the CFTR is defective, then sodium flows freely from the ASL and into the cell. As water follows sodium, the depth of ASL will be depleted and the cilia will be left in the mucous layer.[48] As cilia cannot effectively move in a thick, viscous environment, mucociliary clearance is deficient and a buildup of mucus occurs, clogging small airways.[49] The accumulation of more viscous, nutrient-rich mucus in the lungs allows bacteria to hide from the body's immune system, causing repeated respiratory infections. The presence of the same CFTR proteins in pancreatic duct and skin cells also cause symptoms in these systems.
Chronic infections
The lungs of individuals with cystic fibrosis are colonized and infected by bacteria from an early age. These bacteria, which often spread among individuals with CF, thrive in the altered mucus, which collects in the small airways of the lungs. This mucus leads to the formation of bacterial microenvironments known as biofilms that are difficult for immune cells and antibiotics to penetrate. Viscous secretions and persistent respiratory infections repeatedly damage the lung by gradually remodeling the airways, which makes infection even more difficult to eradicate.[50]
Over time, both the types of bacteria and their individual characteristics change in individuals with CF. In the initial stage, common bacteria such as S. aureus and H. influenzae colonize and infect the lungs.[19] Eventually, Pseudomonas aeruginosa (and sometimes Burkholderia cepacia) dominates. By 18 years of age, 80% of patients with classic CF harbor P. aeruginosa, and 3.5% harbor B. cepacia.[19] Once within the lungs, these bacteria adapt to the environment and develop resistance to commonly used antibiotics. Pseudomonas can develop special characteristics that allow the formation of large colonies, known as "mucoid" Pseudomonas, which are rarely seen in people who do not have CF.[50] Scientific evidences suggest the interleukin 17 pathway plays a key role in resistance and modulation of the inflammatory response during P. aeruginosa infection in CF.[51] In particular, interleukin 17-mediated immunity plays a double-edged activity during chronic airways infection; on one side, it contributes to the control of P. aeruginosa burden, while on the other, it propagates exacerbated pulmonary neutrophilia and tissue remodeling.[51]
Infection can spread by passing between different individuals with CF.[52] In the past, people with CF often participated in summer "CF camps" and other recreational gatherings.[53][54] Hospitals grouped patients with CF into common areas and routine equipment (such as nebulizers)[55] was not sterilized between individual patients.[56] This led to transmission of more dangerous strains of bacteria among groups of patients. As a result, individuals with CF are now routinely isolated from one another in the healthcare setting, and healthcare providers are encouraged to wear gowns and gloves when examining patients with CF to limit the spread of virulent bacterial strains.[57]
CF patients may also have their airways chronically colonized by filamentous fungi (such as Aspergillus fumigatus, Scedosporium apiospermum, Aspergillus terreus) and/or yeasts (such as Candida albicans); other filamentous fungi less commonly isolated include Aspergillus flavus and Aspergillus nidulans (occur transiently in CF respiratory secretions) and Exophiala dermatitidis and Scedosporium prolificans (chronic airway-colonizers); some filamentous fungi such as Penicillium emersonii and Acrophialophora fusispora are encountered in patients almost exclusively in the context of CF.[58] Defective mucociliary clearance characterizing CF is associated with local immunological disorders. In addition, the prolonged therapy with antibiotics and the use of corticosteroid treatments may also facilitate fungal growth. Although the clinical relevance of the fungal airway colonization is still a matter of debate, filamentous fungi may contribute to the local inflammatory response and therefore to the progressive deterioration of the lung function, as often happens with allergic bronchopulmonary aspergillosis – the most common fungal disease in the context of CF, involving a Th2-driven immune response to Aspergillus species.[58][59]
Diagnosis and monitoring

Cystic fibrosis may be diagnosed by many different methods, including newborn screening, sweat testing, and genetic testing.[60] As of 2006 in the United States, 10% of cases are diagnosed shortly after birth as part of newborn screening programs. The newborn screen initially measures for raised blood concentration of immunoreactive trypsinogen.[61] Infants with an abnormal newborn screen need a sweat test to confirm the CF diagnosis. In many cases, a parent makes the diagnosis because the infant tastes salty.[19] Trypsinogen levels can be increased in individuals who have a single mutated copy of the CFTR gene (carriers) or, in rare instances, in individuals with two normal copies of the CFTR gene. Due to these false positives, CF screening in newborns can be controversial.[62][63] Most states and countries do not screen for CF routinely at birth. Therefore, most individuals are diagnosed after symptoms (e.g. sinopulmonary disease and GI manifestations[19]) prompt an evaluation for cystic fibrosis. The most commonly used form of testing is the sweat test. Sweat testing involves application of a medication that stimulates sweating (pilocarpine). To deliver the medication through the skin, iontophoresis is used, whereby one electrode is placed onto the applied medication and an electric current is passed to a separate electrode on the skin. The resultant sweat is then collected on filter paper or in a capillary tube and analyzed for abnormal amounts of sodium and chloride. People with CF have increased amounts of them in their sweat. In contrast, people with CF have less thiocyanate and hypothiocyanite in their saliva[64] and mucus (Banfi et al.). CF can also be diagnosed by identification of mutations in the CFTR gene.[65]
People with CF may be listed in a disease registry that allows researchers and doctors to track health results and identify candidates for clinical trials.[66]
Prenatal
Women who are pregnant or couples planning a pregnancy can have themselves tested for the CFTR gene mutations to determine the risk that their child will be born with CF. Testing is typically performed first on one or both parents and, if the risk of CF is high, testing on the fetus is performed. The American College of Obstetricians and Gynecologists recommends testing for couples who have a personal or close family history of CF, and they recommend that carrier testing be offered to all Caucasian couples and be made available to couples of other ethnic backgrounds.[67]
Because development of CF in the fetus requires each parent to pass on a mutated copy of the CFTR gene and because CF testing is expensive, testing is often performed initially on one parent. If testing shows that parent is a CFTR gene mutation carrier, the other parent is tested to calculate the risk that their children will have CF. CF can result from more than a thousand different mutations.[68] As of 2016, typically only the most common mutations are tested for, such as ΔF508[68] Most commercially available tests look for 32 or fewer different mutations. If a family has a known uncommon mutation, specific screening for that mutation can be performed. Because not all known mutations are found on current tests, a negative screen does not guarantee that a child will not have CF.[69]
During pregnancy, testing can be performed on the placenta (chorionic villus sampling) or the fluid around the fetus (amniocentesis). However, chorionic villus sampling has a risk of fetal death of one in 100 and amniocentesis of one in 200;[70] a recent study has indicated this may be much lower, about one in 1,600.[71]
Economically, for carrier couples of cystic fibrosis, when comparing preimplantation genetic diagnosis (PGD) with natural conception (NC) followed by prenatal testing and abortion of affected pregnancies, PGD provides net economic benefits up to a maternal age around 40 years, after which NC, prenatal testing, and abortion have higher economic benefit.[72]
Management
While no cures for CF are known, several treatment methods are used. The management of CF has improved significantly over the past 70 years. While infants born with it 70 years ago would have been unlikely to live beyond their first year, infants today are likely to live well into adulthood. Recent advances in the treatment of cystic fibrosis have meant that individuals with cystic fibrosis can live a fuller life less encumbered by their condition. The cornerstones of management are the proactive treatment of airway infection, and encouragement of good nutrition and an active lifestyle. Pulmonary rehabilitation as a management of CF continues throughout a person's life, and is aimed at maximizing organ function, and therefore the quality of life. At best, current treatments delay the decline in organ function. Because of the wide variation in disease symptoms, treatment typically occurs at specialist multidisciplinary centers and is tailored to the individual. Targets for therapy are the lungs, gastrointestinal tract (including pancreatic enzyme supplements), the reproductive organs (including assisted reproductive technology), and psychological support.[61]
The most consistent aspect of therapy in CF is limiting and treating the lung damage caused by thick mucus and infection, with the goal of maintaining quality of life. Intravenous, inhaled, and oral antibiotics are used to treat chronic and acute infections. Mechanical devices and inhalation medications are used to alter and clear the thickened mucus. These therapies, while effective, can be extremely time-consuming.
Antibiotics
Many people with CF are on one or more antibiotics at all times, even when healthy, to prophylactically suppress infection. Antibiotics are absolutely necessary whenever pneumonia is suspected or a noticeable decline in lung function is seen, and are usually chosen based on the results of a sputum analysis and the person's past response. This prolonged therapy often necessitates hospitalization and insertion of a more permanent IV such as a peripherally inserted central catheter or Port-a-Cath. Inhaled therapy with antibiotics such as tobramycin, colistin, and aztreonam is often given for months at a time to improve lung function by impeding the growth of colonized bacteria.[73][74][75] Inhaled antibiotic therapy helps lung function by fighting infection, but also has significant drawbacks such as development of antibiotic resistance, tinnitus, and changes in the voice.[76] Oral antibiotics such as ciprofloxacin or azithromycin are given to help prevent infection or to control ongoing infection.[77] The aminoglycoside antibiotics (e.g. tobramycin) used can cause hearing loss, damage to the balance system in the inner ear or renal failure with long-term use.[78] To prevent these side-effects, the amount of antibiotics in the blood is routinely measured and adjusted accordingly.
Other treatments for lung disease
Several mechanical techniques are used to dislodge sputum and encourage its expectoration. In the hospital setting, chest physiotherapy is used; a respiratory therapist percusses an individual's chest by hand several times a day, to loosen up secretions. Devices that recreate this percussive therapy include the ThAIRapy Vest and the intrapulmonary percussive ventilator. Newer methods such as biphasic cuirass ventilation, and associated clearance mode available in such devices, integrate a cough assistance phase, as well as a vibration phase for dislodging secretions. These are portable and adapted for home use.[79]
Ivacaftor is an oral medication for the treatment of CF due to a number of specific mutations.[80][81] It improves lung function by about 10%; however, as of 2014 is expensive.[80]
Aerosolized medications that help loosen secretions include dornase alfa and hypertonic saline.[82] Dornase is a recombinant human deoxyribonuclease, which breaks down DNA in the sputum, thus decreasing its viscosity.[83] Denufosol, an investigational drug, opens an alternative chloride channel, helping to liquefy mucus.[84] Whether inhaled corticosteroids are usefulis unclear.[85]
As lung disease worsens, mechanical breathing support may become necessary. Individuals with CF may need to wear special masks at night to help push air into their lungs. These machines, known as bilevel positive airway pressure (BiPAP) ventilators, help prevent low blood oxygen levels during sleep. BiPAP may also be used during physical therapy to improve sputum clearance.[86] During severe illness, a tube may be placed in the throat (a procedure known as a tracheostomy) to enable breathing supported by a ventilator.
For children, preliminary studies show massage therapy may help people and their families' quality of life.[87] Pneumococcal vaccination has not been studied as of 2014.[88]
Some lung infections require surgical removal of the infected part of the lung. If this is necessary many times, lung function is severely reduced.[89]
Transplantation
Lung transplantation often becomes necessary for individuals with CF as lung function and exercise tolerance decline. Although single lung transplantation is possible in other diseases, individuals with CF must have both lungs replaced because the remaining lung might contain bacteria that could infect the transplanted lung. A pancreatic or liver transplant may be performed at the same time to alleviate liver disease and/or diabetes.[90] Lung transplantation is considered when lung function declines to the point where assistance from mechanical devices is required or someone's survival is threatened.[91]
Other aspects

Newborns with intestinal obstruction typically require surgery, whereas adults with distal intestinal obstruction syndrome typically do not. Treatment of pancreatic insufficiency by replacement of missing digestive enzymes allows the duodenum to properly absorb nutrients and vitamins that would otherwise be lost in the feces. However, the best dosage and form of pancreatic enzyme replacement is unclear, as are the risks and long-term effectiveness of this treatment.[92]
So far, no large-scale research involving the incidence of atherosclerosis and coronary heart disease in adults with cystic fibrosis has been conducted. This is likely because the vast majority of people with cystic fibrosis do not live long enough to develop clinically significant atherosclerosis or coronary heart disease.
Diabetes is the most common nonpulmonary complication of CF. It mixes features of type 1 and type 2 diabetes, and is recognized as a distinct entity, cystic fibrosis-related diabetes.[33][93] While oral antidiabetic drugs are sometimes used, the only recommended treatment is the use of insulin injections or an insulin pump,[94] and, unlike in type 1 and 2 diabetes, dietary restrictions are not recommended.[33]
Development of osteoporosis can be prevented by increased intake of vitamin D and calcium, and can be treated by bisphosphonates, although adverse effects can be an issue.[95] Poor growth may be avoided by insertion of a feeding tube for increasing food energy through supplemental feeds or by administration of injected growth hormone.[96]
Sinus infections are treated by prolonged courses of antibiotics. The development of nasal polyps or other chronic changes within the nasal passages may severely limit airflow through the nose, and over time reduce the person's sense of smell. Sinus surgery is often used to alleviate nasal obstruction and to limit further infections. Nasal steroids such as fluticasone are used to decrease nasal inflammation.[97]
Female infertility may be overcome by assisted reproduction technology, particularly embryo transfer techniques. Male infertility caused by absence of the vas deferens may be overcome with testicular sperm extraction, collecting sperm cells directly from the testicles. If the collected sample contains too few sperm cells to likely have a spontaneous fertilization, intracytoplasmic sperm injection can be performed.[98] Third party reproduction is also a possibility for women with CF. Whether taking antioxidants affects outcomes is unclear.[99]
Prognosis
The prognosis for cystic fibrosis has improved due to earlier diagnosis through screening and better treatment and access to health care. In 1959, the median age of survival of children with CF in the United States was six months.[100] In 2010, survival is estimated to be 37 years for women and 40 for men.[101] In Canada, median survival increased from 24 years in 1982 to 47.7 in 2007.[102]
Of those with CF who are more than 18 years old as of 2009, 92% had graduated from high school, 67% had at least some college education, 15% were disabled, 9% were unemployed, 56% were single, and 39% were married or living with a partner.[103]
Quality of life
Chronic illnesses can be very difficult to manage. CF is a chronic illness that affects the "digestive and respiratory tracts resulting in generalized malnutrition and chronic respiratory infections".[104] The thick secretions clog the airways in the lungs, which often cause inflammation and severe lung infections.[105][106] If it is compromised, it affects the quality of life of someone with CF and their ability to complete such tasks as everyday chores. It is important for CF patients to understand the detrimental relationship that chronic illnesses place on the quality of life (QOL). According to Schmitz and Goldbeck (2006), the fact that CF significantly increases emotional stress on both the individual and the family, "and the necessary time-consuming daily treatment routine may have further negative effects on quality of life".[107] However, Havermans and colleagues (2006) have shown that young outpatients with CF who have participated in the Cystic Fibrosis Questionnaire-Revised "rated some QOL domains higher than did their parents".[108] Consequently, outpatients with CF have a more positive outlook for themselves. Furthermore, many ways can improve the QOL in CF patients. Exercise is promoted to increase lung function. Integrating an exercise regimen into the CF patient’s daily routine can significantly improve QOL.[109] No definitive cure for CF is known, but diverse medications are used, such as mucolytics, bronchodilators, steroids, and antibiotics, that have the purpose of loosening mucus, expanding airways, decreasing inflammation, and fighting lung infections, respectively.[110]
Epidemiology
| Mutation | Frequency worldwide[111] |
|---|---|
| ΔF508 | 66%–70%[19] |
| G542X | 2.4% |
| G551D | 1.6% |
| N1303K | 1.3% |
| W1282X | 1.2% |
| All others | 27.5% |
Cystic fibrosis is the most common life-limiting autosomal recessive disease among people of European heritage.[112] In the United States, about 30,000 individuals have CF; most are diagnosed by six months of age. In Canada, about 4,000 people have CF.[113] Around one 1 in 25 people of European descent, and one in 30 of Caucasian Americans,[114] is a carrier of a CF mutation. Although CF is less common in these groups, roughly one in 46 Hispanics, one in 65 Africans, and one in 90 Asians carry at least one abnormal CFTR gene.[115][116] Ireland has the world's highest prevalence of CF, at one in 1353.[117]
Although technically a rare disease, CF is ranked as one of the most widespread life-shortening genetic diseases. It is most common among nations in the Western world. An exception is Finland, where only one in 80 people carries a CF mutation.[118] The World Health Organization states, "In the European Union, one in 2000–3000 newborns is found to be affected by CF".[119] In the United States, one in 3,500 children is born with CF.[120] In 1997, about one in 3,300 Caucasian children in the United States was born with CF. In contrast, only one in 15,000 African American children suffered from it, and in Asian Americans, the rate was even lower at one in 32,000.[121]
Cystic fibrosis is diagnosed in males and females equally. For reasons that remain unclear, data have shown that males tend to have a longer life expectancy than females,[122][123] but recent studies suggest this gender gap may no longer exist perhaps due to improvements in health care facilities,[124][125] while a recent study from Ireland identified a link between the female hormone estrogen and worse outcomes in CF.[126]
The distribution of CF alleles varies among populations. The frequency of ΔF508 carriers has been estimated at one in 200 in northern Sweden, one in 143 in Lithuanians, and one in 38 in Denmark. No ΔF508 carriers were found among 171 Finns and 151 Saami people.[127] ΔF508 does occur in Finland, but it is a minority allele there. CF is known to occur in only 20 families (pedigrees) in Finland.[128]
Evolution
The ΔF508 mutation is estimated to be up to 52,000 years old.[129] Numerous hypotheses have been advanced as to why such a lethal mutation has persisted and spread in the human population. Other common autosomal recessive diseases such as sickle-cell anemia have been found to protect carriers from other diseases, a concept known as heterozygote advantage. Resistance to the following have all been proposed as possible sources of heterozygote advantage:
- Cholera: With the discovery that cholera toxin requires normal host CFTR proteins to function properly, it was hypothesized that carriers of mutant CFTR genes benefited from resistance to cholera and other causes of diarrhea.[130] Further studies have not confirmed this hypothesis.[131][132]
- Typhoid: Normal CFTR proteins are also essential for the entry of Salmonella Typhi into cells,[133] suggesting that carriers of mutant CFTR genes might be resistant to typhoid fever. No in vivo study has yet confirmed this. In both cases, the low level of cystic fibrosis outside of Europe, in places where both cholera and typhoid fever are endemic, is not immediately explicable.
- Diarrhea: The prevalence of CF in Europe might be connected with the development of cattle domestication. In this hypothesis, carriers of a single mutant CFTR had some protection from diarrhea caused by lactose intolerance, prior to the appearance of the mutations that created lactose tolerance.[134]
- Tuberculosis: Another possible explanation is that carriers of the gene could have some resistance to TB.[135][136] This hypothesis is based on the thesis that CFTR gene mutation carriers have insufficient action in one of their enzymes – arylsulphatase - which is necessary for Mycobacterium tuberculosis virulence. As M. tuberculosis would use its host’s sources to affect the individual, and due to the lack of enzyme it could not presents its virulence, being a carrier of CFTR mutation could provide resistance against tuberculosis.[137]
History

CF is supposed to have appeared about 3,000 BC because of migration of peoples, gene mutations, and new conditions in nourishment.[138] Although the entire clinical spectrum of CF was not recognized until the 1930s, certain aspects of CF were identified much earlier. Indeed, literature from Germany and Switzerland in the 18th century warned "Wehe dem Kind, das beim Kuß auf die Stirn salzig schmeckt, es ist verhext und muss bald sterben" or "Woe to the child who tastes salty from a kiss on the brow, for he is cursed and soon must die", recognizing the association between the salt loss in CF and illness.[138]
In the 19th century, Carl von Rokitansky described a case of fetal death with meconium peritonitis, a complication of meconium ileus associated with CF. Meconium ileus was first described in 1905 by Karl Landsteiner.[138] In 1936, Guido Fanconi described a connection between celiac disease, cystic fibrosis of the pancreas, and bronchiectasis.[139]
In 1938, Dorothy Hansine Andersen published an article, "Cystic Fibrosis of the Pancreas and Its Relation to Celiac Disease: a Clinical and Pathological Study", in the American Journal of Diseases of Children. She was the first to describe the characteristic cystic fibrosis of the pancreas and to correlate it with the lung and intestinal disease prominent in CF.[9] She also first hypothesized that CF was a recessive disease and first used pancreatic enzyme replacement to treat affected children. In 1952, Paul di Sant’Agnese discovered abnormalities in sweat electrolytes; a sweat test was developed and improved over the next decade.[140]
The first linkage between CF and another marker (Paroxonase) was found in 1985 by Hans Eiberg, indicating that only one locus exists for CF. In 1988, the first mutation for CF, ΔF508 was discovered by Francis Collins, Lap-Chee Tsui, and John R. Riordan on the seventh chromosome. Subsequent research has found over 1,000 different mutations that cause CF.
Because mutations in the CFTR gene are typically small, classical genetics techniques had been unable to accurately pinpoint the mutated gene.[141] Using protein markers, gene-linkage studies were able to map the mutation to chromosome 7. Chromosome-walking and -jumping techniques were then used to identify and sequence the gene.[142] In 1989, Lap-Chee Tsui led a team of researchers at the Hospital for Sick Children in Toronto that discovered the gene responsible for CF. CF represents a classic example of how a human genetic disorder was elucidated strictly by the process of forward genetics.
Research
Gene therapy
Gene therapy has been explored as a potential cure for CF. Results from trials have shown limited success as of 2013.[143] A small study published in 2015 found a small benefit.[144]
The focus of much CF gene therapy research is aimed at trying to place a normal copy of the CFTR gene into affected cells. Transferring the normal CFTR gene into the affected epithelium cells would result in the production of functional CFTR protein in all target cells, without adverse reactions or an inflammation response. To prevent the lung manifestations of CF, only 5–10% the normal amount of CFTR gene expression is needed.[145] Multiple approaches have been tested for gene transfer, such as liposomes and viral vectors in animal models and clinical trials. However, both methods were found to be relatively inefficient treatment options,[146] mainly because very few cells take up the vector and express the gene, so the treatment has little effect. Additionally, problems have been noted in cDNA recombination, such that the gene introduced by the treatment is rendered unusable.[147] There has been a functional repair in culture of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients.[148]
Small molecules
A number of small molecules that aim at compensating various mutations of the CFTR gene are under development. One approach is to develop drugs that get the ribosome to overcome the stop codon and synthesize a full-length CFTR protein. About 10% of CF results from a premature stop codon in the DNA, leading to early termination of protein synthesis and truncated proteins. These drugs target nonsense mutations such as G542X, which consists of the amino acid glycine in position 542 being replaced by a stop codon. Aminoglycoside antibiotics interfere with protein synthesis and error-correction. In some cases, they can cause the cell to overcome a premature stop codon by inserting a random amino acid, thereby allowing expression of a full-length protein.[149] The aminoglycoside gentamicin has been used to treat lung cells from CF patients in the laboratory to induce the cells to grow full-length proteins.[150] Another drug targeting nonsense mutations is ataluren, which is undergoing Phase III clinical trials as of October 2011.[151]
Other
It is unclear as of 2014 if ursodeoxycholic acid is useful for those with cystic fibrosis-related liver disease.[152]
Society and culture
- Sick: The Life and Death of Bob Flanagan, Supermasochist
- 65_Redroses
- Breathing for a Living, a memoir by Laura Rothenberg
References
- 1 2 3 4 5 6 7 8 9 O'Sullivan, BP; Freedman, SD (30 May 2009). "Cystic fibrosis.". Lancet. 373 (9678): 1891–904. doi:10.1016/s0140-6736(09)60327-5. PMID 19403164.
- 1 2 3 Hodson, Margaret; Geddes, Duncan; Bush, Andrew, eds. (2012). Cystic fibrosis (3rd ed.). London: Hodder Arnold. p. 3. ISBN 978-1-4441-1369-3.
- 1 2 3 Massie, J; Delatycki, MB (December 2013). "Cystic fibrosis carrier screening.". Paediatric respiratory reviews. 14 (4): 270–5. doi:10.1016/j.prrv.2012.12.002. PMID 23466339.
- ↑ Buckingham, Lela (2012). Molecular diagnostics fundamentals, methods, and clinical applications (2nd ed.). Philadelphia: F.A. Davis Co. p. 351. ISBN 978-0-8036-2975-2.
- ↑ Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D (2004). "Cystic fibrosis adult care consensus conference report". Chest. 125 (90010): 1–39. doi:10.1378/chest.125.1_suppl.1S. PMID 14734689.
- ↑ Warnock, L; Gates, A (21 December 2015). "Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis.". The Cochrane database of systematic reviews (12): CD001401. PMID 26688006.
- ↑ Ong, T; Ramsey, BW (15 September 2015). "Update in Cystic Fibrosis 2014.". American Journal of Respiratory and Critical Care Medicine. 192 (6): 669–75. doi:10.1164/rccm.201504-0656UP. PMID 26371812.
- ↑ Nazareth, D; Walshaw, M (October 2013). "Coming of age in cystic fibrosis - transition from paediatric to adult care.". Clinical medicine (London, England). 13 (5): 482–6. doi:10.7861/clinmedicine.13-5-482. PMID 24115706.
- 1 2 Andersen DH (1938). "Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study". Am J Dis Child. 56: 344–399. doi:10.1001/archpedi.1938.01980140114013.
- ↑ Quinton PM (June 2007). "Cystic fibrosis: lessons from the sweat gland". Physiology (Bethesda). 22 (3): 212–25. doi:10.1152/physiol.00041.2006. PMID 17557942.
- 1 2 Hardin DS (August 2004). "GH improves growth and clinical status in children with cystic fibrosis – a review of published studies". Eur. J. Endocrinol. 151 (Suppl 1): S81–5. doi:10.1530/eje.0.151S081. PMID 15339250.
- 1 2 De Lisle RC (September 2009). "Pass the bicarb: the importance of HCO3- for mucin release". J. Clin. Invest. 119 (9): 2535–7. doi:10.1172/JCI40598. PMC 2735941
 . PMID 19726878.
. PMID 19726878. - ↑ O'Malley CA (May 2009). "Infection control in cystic fibrosis: cohorting, cross-contamination, and the respiratory therapist" (PDF). Respir Care. 54 (5): 641–57. doi:10.4187/aarc0446. PMID 19393108.
- ↑ Makker K, Agarwal A, Sharma R (April 2009). "Oxidative stress & male infertility" (PDF). Indian J. Med. Res. 129 (4): 357–67. PMID 19535829.
- ↑ Blackman SM, Deering-Brose R, McWilliams R, Naughton K, Coleman B, Lai T, Algire M, Beck S, Hoover-Fong J, Hamosh A, Fallin MD, West K, Arking DE, Chakravarti A, Cutler DJ, Cutting GR (October 2006). "Relative contribution of genetic and nongenetic modifiers to intestinal obstruction in cystic fibrosis". Gastroenterology. 131 (4): 1030–9. doi:10.1053/j.gastro.2006.07.016. PMC 1764617. PMID 17030173.
- ↑ Ratjen FA (May 2009). "Cystic fibrosis: pathogenesis and future treatment strategies" (PDF). Respir Care. 54 (5): 595–605. doi:10.4187/aarc0427. PMID 19393104.
- ↑ Reaves J, Wallace G (2010). "Unexplained bruising: weighing the pros and cons of possible causes". Consultant for Pediatricians. 9: 201–2.
- ↑ Flume PA, Mogayzel Jr PJ, Robinson KA, et al. (March 2010). "Cystic Fibrosis Pulmonary Guidelines: Pulmonary Complications: Hemoptysis and Pneumthorax". Am J Respir Crit Care Med. 182 (3): 298–306. doi:10.1164/rccm.201002-0157OC. PMID 20299528.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 Mitchell, Richard Sheppard; Kumar, Vinay; Robbins, Stanley L.; Abbas, Abul K.; Fausto, Nelson (2007). Robbins basic pathology. Saunders/Elsevier. ISBN 1-4160-2973-7.
- 1 2 3 4 Rowe SM, Miller S, Sorscher EJ (May 2005). "Cystic fibrosis". The New England Journal of Medicine. 352 (19): 1992–2001. doi:10.1056/NEJMra043184. PMID 15888700.
- ↑ Girón RM, Domingo D, Buendía B, Antón E, Ruiz-Velasco LM, Ancochea J (October 2005). "Nontuberculous mycobacteria in patients with cystic fibrosis". Arch. Bronconeumol. (in Spanish). 41 (10): 560–5. doi:10.1016/S1579-2129(06)60283-8. PMID 16266669.
- ↑ Franco LP, Camargos PA, Becker HM, Guimarães RE (December 2009). "Nasal endoscopic evaluation of children and adolescents with cystic fibrosis". Braz J Otorhinolaryngol. 75 (6): 806–13. doi:10.1590/S1808-86942009000600006. PMID 20209279.
- ↑ Maldonado M, Martínez A, Alobid I, Mullol J (December 2004). "The antrochoanal polyp". Rhinology. 42 (4): 178–82. PMID 15626248.
- ↑ Ramsey B, Richardson MA (September 1992). "Impact of sinusitis in cystic fibrosis". J. Allergy Clin. Immunol. 90 (3 Pt 2): 547–52. doi:10.1016/0091-6749(92)90183-3. PMID 1527348.
- ↑ Eggermont E, De Boeck K (October 1991). "Small-intestinal abnormalities in cystic fibrosis patients". Eur. J. Pediatr. 150 (12): 824–8. doi:10.1007/BF01954999. PMID 1743211.
- ↑ Kulczycki LL, Shwachman H (August 1958). "Studies in cystic fibrosis of the pancreas; occurrence of rectal prolapse". N. Engl. J. Med. 259 (9): 409–12. doi:10.1056/NEJM195808282590901. PMID 13578072.
- ↑ Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS (September 1998). "Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis". N. Engl. J. Med. 339 (10): 653–8. doi:10.1056/NEJM199809033391002. PMID 9725922.
- ↑ Malfroot A, Dab I (November 1991). "New insights on gastro-esophageal reflux in cystic fibrosis by longitudinal follow up". Arch. Dis. Child. 66 (11): 1339–45. doi:10.1136/adc.66.11.1339. PMC 1793275. PMID 1755649.
- ↑ Khoshoo V, Udall JN (February 1994). "Meconium ileus equivalent in children and adults". Am. J. Gastroenterol. 89 (2): 153–7. PMID 8304294.
- ↑ Williams SG, Westaby D, Tanner MS, Mowat AP (October 1992). "Liver and biliary problems in cystic fibrosis". Br. Med. Bull. 48 (4): 877–92. PMID 1458306.
- ↑ Colombo C, Russo MC, Zazzeron L, Romano G (July 2006). "Liver disease in cystic fibrosis". J. Pediatr. Gastroenterol. Nutr. 43 (Suppl 1): S49–55. doi:10.1097/01.mpg.0000226390.02355.52. PMID 16819402.
- ↑ Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, Adams KS, Seaquist ER (August 1994). "Insulin sensitivity in cystic fibrosis". Diabetes. 43 (8): 1020–6. doi:10.2337/diabetes.43.8.1020. PMID 8039595.
- 1 2 3 Alves Cde A, Aguiar RA, Alves AC, Santana MA (April 2007). "Diabetes mellitus in patients with cystic fibrosis". J Bras Pneumol. 33 (2): 213–21. doi:10.1590/S1806-37132007000200017. PMID 17724542.
- ↑ Haworth CS, Selby PL, Webb AK, Dodd ME, Musson H, McL Niven R, Economou G, Horrocks AW, Freemont AJ, Mawer EB, Adams JE (November 1999). "Low bone mineral density in adults with cystic fibrosis". Thorax. 54 (11): 961–7. doi:10.1136/thx.54.11.961. PMC 1745400. PMID 10525552.
- ↑ Vandemergel X, Decaux G (April 2003). "[Review on hypertrophic osteoarthropathy and digital clubbing]". Revue Médicale de Bruxelles (in French). 24 (2): 88–94. PMID 12806875.
- ↑ Pitts-Tucker TJ, Miller MG, Littlewood JM (June 1986). "Finger clubbing in cystic fibrosis". Arch. Dis. Child. 61 (6): 576–9. doi:10.1136/adc.61.6.576. PMC 1777828. PMID 3488032.
- ↑ McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD (October 2000). "Fertility in men with cystic fibrosis: an update on current surgical practices and outcomes". Chest. 118 (4): 1059–62. doi:10.1378/chest.118.4.1059. PMID 11035677.
- ↑ Chen H, Ruan YC, Xu WM, Chen J, Chan HC (2012). "Regulation of male fertility by CFTR and implications in male infertility". Human Reproduction Update. 18 (6): 703–713. doi:10.1093/humupd/dms027. PMID 22709980.
- ↑ Augarten A, Yahav Y, Kerem BS, Halle D, Laufer J, Szeinberg A, Dor J, Mashiach S, Gazit E, Madgar I (November 1994). "Congenital bilateral absence of vas deferens in the absence of cystic fibrosis". Lancet. 344 (8935): 1473–4. doi:10.1016/S0140-6736(94)90292-5. PMID 7968122.
- ↑ Gilljam M, Antoniou M, Shin J, Dupuis A, Corey M, Tullis DE (July 2000). "Pregnancy in cystic fibrosis. Fetal and maternal outcome". Chest. 118 (1): 85–91. doi:10.1378/chest.118.1.85. PMID 10893364.
- ↑ "Profile : Lap-Chee Tsui". Science.ca. 1989-05-09. Retrieved 2013-01-23.
- ↑ Bobadilla JL, Macek M, Fine JP, Farrell PM (June 2002). "Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening". Hum. Mutat. 19 (6): 575–606. doi:10.1002/humu.10041. PMID 12007216.
- ↑ Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, Stutts MJ, Milgram SL (July 1998). "An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton". J. Biol. Chem. 273 (31): 19797–801. doi:10.1074/jbc.273.31.19797. PMID 9677412.
- ↑ Childers, Eckel & Himmel 2007
- ↑ Xu, Szép & Lu 2009
- ↑ Moskwa, Lorentzen & Excoffon 2007
- ↑ Conner, Wijkstrom-Frei & Randell 2007
- ↑ Verkman AS, Song Y, Thiagarajah JR. Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease. Am J Physiol Cell Physiol. 2003;284(1):C2–C15
- ↑ Marieb & Hoehn, (2014) Human Anatomy and Physiology, Chapter 22: The Respiratory System, pg 906, Pearson Education
- 1 2 Saiman L (2004). "Microbiology of early CF lung disease". Paediatric Respiratory Reviews. 5 (Suppl A): S367–69. doi:10.1016/S1526-0542(04)90065-6. PMID 14980298.
- 1 2 Lorè NI, Cigana C, Riva C, De Fino I, et al. (May 2016). "IL-17A impairs host tolerance during airway chronic infection by Pseudomonas aeruginosa". Scientific Reports. 6 :25937: 25937. doi:10.1038/srep25937. PMC 4870500. PMID 27189736.
- ↑ Tümmler B, Koopmann U, Grothues D, Weissbrodt H, Steinkamp G, von der Hardt H (June 1991). "Nosocomial acquisition of Pseudomonas aeruginosa by cystic fibrosis patients". J. Clin. Microbiol. 29 (6): 1265–7. Bibcode:1991JPoSA..29.1265A. doi:10.1002/pola.1991.080290905. PMC 271975. PMID 1907611.
- ↑ "Pseudomonas cepacia at summer camps for persons with cystic fibrosis". MMWR Morb. Mortal. Wkly. Rep. 42 (23): 456–9. June 1993. PMID 7684813.
- ↑ Pegues DA, Carson LA, Tablan OC, FitzSimmons SC, Roman SB, Miller JM, Jarvis WR (May 1994). "Acquisition of Pseudomonas cepacia at summer camps for patients with cystic fibrosis. Summer Camp Study Group". J. Pediatr. 124 (5 Pt 1): 694–702. doi:10.1016/S0022-3476(05)81357-5. PMID 7513755.
- ↑ Pankhurst CL, Philpott-Howard J (April 1996). "The environmental risk factors associated with medical and dental equipment in the transmission of Burkholderia (Pseudomonas) cepacia in cystic fibrosis patients". J. Hosp. Infect. 32 (4): 249–55. doi:10.1016/S0195-6701(96)90035-3. PMID 8744509.
- ↑ Jones AM, Govan JR, Doherty CJ, Dodd ME, Isalska BJ, Stanbridge TN, Webb AK (June 2003). "Identification of airborne dissemination of epidemic multiresistant strains of Pseudomonas aeruginosa at a CF centre during a cross infection outbreak". Thorax. 58 (6): 525–27. doi:10.1136/thorax.58.6.525. PMC 1746694. PMID 12775867.
- ↑ Høiby N (June 1995). "Isolation and treatment of cystic fibrosis patients with lung infections caused by Pseudomonas (Burkholderia) cepacia and multiresistant Pseudomonas aeruginosa". Neth J Med. 46 (6): 280–87. doi:10.1016/0300-2977(95)00020-N. PMID 7643943.
- 1 2 Pihet M, Carrere J, Cimon B, Chabasse D, Delhaes L, Symoens F, Bouchara JP (June 2009). "Occurrence and relevance of filamentous fungi in respiratory secretions of patients with cystic fibrosis—a review". Med Mycol. 47 (4): 387–97. doi:10.1080/13693780802609604. PMID 19107638.
- ↑ Rapaka RR, Kolls JK (2009). "Pathogenesis of allergic bronchopulmonary aspergillosis in cystic fibrosis: current understanding and future directions". Med Mycol. 47 (Suppl 1): S331–7. doi:10.1080/13693780802266777. PMID 18668399.
- ↑ Mishra A, Greaves R, Massie J (November 2005). "The relevance of sweat testing for the diagnosis of cystic fibrosis in the genomic era.". The Clinical biochemist. Reviews / Australian Association of Clinical Biochemists. 26 (4): 135–53. PMC 1320177. PMID 16648884.
- 1 2 Davies JC, Alton EW, Bush A (December 2007). "Cystic fibrosis". BMJ. 335 (7632): 1255–9. doi:10.1136/bmj.39391.713229.AD. PMC 2137053. PMID 18079549.
- ↑ Ross LF (September 2008). "Newborn screening for cystic fibrosis: a lesson in public health disparities". The Journal of Pediatrics. 153 (3): 308–13. doi:10.1016/j.jpeds.2008.04.061. PMC 2569148. PMID 18718257.
- ↑ Assael BM, Castellani C, Ocampo MB, Iansa P, Callegaro A, Valsecchi MG (September 2002). "Epidemiology and survival analysis of cystic fibrosis in an area of intense neonatal screening over 30 years". American Journal of Epidemiology. 156 (5): 397–401. doi:10.1093/aje/kwf064. PMID 12196308.
- ↑ Minarowski, Sands & Minarowska 2008
- ↑ Stern RC (February 1997). "The diagnosis of cystic fibrosis". N. Engl. J. Med. 336 (7): 487–91. doi:10.1056/NEJM199702133360707. PMID 9017943.
- ↑ Freudenheim, Milt (2009-12-22). "Tool in Cystic Fibrosis Fight: A Registry". New York Times. pp. D1. Retrieved 2009-12-21.
- ↑ American College of Obstetricians and Gynecologists; American College of Medical Genetics (2001). Preconception and prenatal carrier screening for cystic fibrosis. Clinical and laboratory guidelines. Washington DC: American College of Obstetricians and Gynecologists. ISBN 0-915473-74-7.
- 1 2 Elborn, JS (29 April 2016). "Cystic fibrosis.". Lancet (London, England). doi:10.1016/S0140-6736(16)00576-6. PMID 27140670.
- ↑ Elias S, Annas GJ, Simpson JL (April 1991). "Carrier screening for cystic fibrosis: implications for obstetric and gynecologic practice". Am. J. Obstet. Gynecol. 164 (4): 1077–83. doi:10.1016/0002-9378(91)90589-j. PMID 2014829.
- ↑ Tabor A, Philip J, Madsen M, Bang J, Obel EB, Nørgaard-Pedersen B (June 1986). "Randomised controlled trial of genetic amniocentesis in 4606 low-risk women". Lancet. 1 (8493): 1287–93. doi:10.1016/S0140-6736(86)91218-3. PMID 2423826.
- ↑ Eddleman KA, Malone FD, Sullivan L, Dukes K, Berkowitz RL, Kharbutli Y, Porter TF, Luthy DA, Comstock CH, Saade GR, Klugman S, Dugoff L, Craigo SD, Timor-Tritsch IE, Carr SR, Wolfe HM, D'Alton ME (November 2006). "Pregnancy loss rates after midtrimester amniocentesis". Obstet Gynecol. 108 (5): 1067–72. doi:10.1097/01.AOG.0000240135.13594.07. PMID 17077226.
- ↑ Davis LB, Champion SJ, Fair SO, Baker VL, Garber AM (April 2010). "A cost-benefit analysis of preimplantation genetic diagnosis for carrier couples of cystic fibrosis". Fertil. Steril. 93 (6): 1793–804. doi:10.1016/j.fertnstert.2008.12.053. PMID 19439290.
- ↑ Pai VB, Nahata MC (October 2001). "Efficacy and safety of aerosolized tobramycin in cystic fibrosis". Pediatr. Pulmonol. 32 (4): 314–27. doi:10.1002/ppul.1125. PMID 11568993.
- ↑ Westerman EM, Le Brun PP, Touw DJ, Frijlink HW, Heijerman HG (March 2004). "Effect of nebulized colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis: a pilot study". J. Cyst. Fibros. 3 (1): 23–8. doi:10.1016/j.jcf.2003.12.005. PMID 15463883.
- ↑ McCoy KS, Quittner AL, Oermann CM, Gibson RL, Retsch-Bogart GZ, Montgomery AB (November 2008). "Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis". Am. J. Respir. Crit. Care Med. 178 (9): 921–8. doi:10.1164/rccm.200712-1804OC. PMC 2577727. PMID 18658109.
- ↑ Ryan G, Singh M, Dwan K (2011). "Inhaled antibiotics for long-term therapy in cystic fibrosis". The Cochrane Database of Systematic Reviews (3): CD001021. doi:10.1002/14651858.CD001021.pub2. PMID 21412868.
- ↑ Hansen CR, Pressler T, Koch C, Høiby N (March 2005). "Long-term azitromycin treatment of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection; an observational cohort study". J. Cyst. Fibros. 4 (1): 35–40. doi:10.1016/j.jcf.2004.09.001. PMID 15752679.
- ↑ Tan KH, Mulheran M, Knox AJ, Smyth AR (March 2003). "Aminoglycoside prescribing and surveillance in cystic fibrosis". Am. J. Respir. Crit. Care Med. 167 (6): 819–23. doi:10.1164/rccm.200109-012CC. PMID 12623858.
- ↑ van der Schans C, Prasad A, Main E (2000). Van Der Schans CP, ed. "Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis". Cochrane Database Syst Rev (2): CD001401. doi:10.1002/14651858.CD001401. PMID 10796781.
- 1 2 Whiting, P; Al, M; Burgers, L; Westwood, M; Ryder, S; Hoogendoorn, M; Armstrong, N; Allen, A; Severens, H; Kleijnen, J (March 2014). "Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis.". Health technology assessment (Winchester, England). 18 (18): 1–106. doi:10.3310/hta18180. PMID 24656117.
- ↑ Wainwright, CE (October 2014). "Ivacaftor for patients with cystic fibrosis.". Expert review of respiratory medicine. 8 (5): 533–8. doi:10.1586/17476348.2014.951333. PMID 25148205.
- ↑ Kuver R, Lee SP (April 2006). "Hypertonic saline for cystic fibrosis". N. Engl. J. Med. 354 (17): 1848–51; author reply 1848–51. doi:10.1056/NEJMc060351. PMID 16642591.
- ↑ Lieberman J (July 1968). "Dornase aerosol effect on sputum viscosity in cases of cystic fibrosis". JAMA. 205 (5): 312–3. doi:10.1001/jama.205.5.312. PMID 5694947.
- ↑ Kellerman D, Rossi Mospan A, Engels J, Schaberg A, Gorden J, Smiley L (2008). "Denufosol: a review of studies with inhaled P2Y(2) agonists that led to Phase 3". Pulmonary Pharmacology & Therapeutics. 21 (4): 600–7. doi:10.1016/j.pupt.2007.12.003. PMID 18276176.
- ↑ Balfour-Lynn, IM; Welch, K (Oct 9, 2014). "Inhaled corticosteroids for cystic fibrosis.". The Cochrane database of systematic reviews. 10: CD001915. doi:10.1002/14651858.CD001915.pub4. PMID 25300165.
- ↑ Moran F, Bradley JM, Piper AJ (2009). Moran F, ed. "Non-invasive ventilation for cystic fibrosis". Cochrane Database Syst Rev (1): CD002769. doi:10.1002/14651858.CD002769.pub3. PMID 19160211.
- ↑ Huth MM, Zink KA, Van Horn NR (2005). "The effects of massage therapy in improving outcomes for youth with cystic fibrosis: an evidence review". Pediatr Nurs. 31 (4): 328–32. PMID 16229132.
- ↑ Burgess, L; Southern, KW (Aug 5, 2014). "Pneumococcal vaccines for cystic fibrosis.". The Cochrane database of systematic reviews. 8: CD008865. doi:10.1002/14651858.CD008865.pub3. PMID 25093421.
- ↑ http://emedicine.medscape.com/article/906209-overview#a2
- ↑ Fridell JA, Vianna R, Kwo PY, Howenstine M, Sannuti A, Molleston JP, Pescovitz MD, Tector AJ (October 2005). "Simultaneous liver and pancreas transplantation in patients with cystic fibrosis". Transplant. Proc. 37 (8): 3567–9. doi:10.1016/j.transproceed.2005.09.091. PMID 16298663.
- ↑ Belkin RA, Henig NR, Singer LG, Chaparro C, Rubenstein RC, Xie SX, Yee JY, Kotloff RM, Lipson DA, Bunin GR (March 2006). "Risk factors for death of patients with cystic fibrosis awaiting lung transplantation". Am. J. Respir. Crit. Care Med. 173 (6): 659–66. doi:10.1164/rccm.200410-1369OC. PMC 2662949. PMID 16387803.
- ↑ Somaraju, UR; Solis-Moya, A (Oct 13, 2014). "Pancreatic enzyme replacement therapy for people with cystic fibrosis.". The Cochrane database of systematic reviews. 10: CD008227. doi:10.1002/14651858.CD008227.pub2. PMID 25310479.
- ↑ Zirbes J, Milla CE (September 2009). "Cystic fibrosis related diabetes". Paediatr Respir Rev. 10 (3): 118–23; quiz 123. doi:10.1016/j.prrv.2009.04.004. PMID 19651382.
- ↑ Onady GM, Stolfi A (2005). Onady, Gary M, ed. "Insulin and oral agents for managing cystic fibrosis-related diabetes". Cochrane Database Syst Rev (3): CD004730. doi:10.1002/14651858.CD004730.pub2. PMID 16034943.
- ↑ Conwell LS, Chang AB (2012). Conwell, Louise S, ed. "Bisphosphonates for osteoporosis in people with cystic fibrosis". Cochrane Database Syst Rev. 4 (4): CD002010. doi:10.1002/14651858.CD002010.pub3. PMID 22513903.
- ↑ Hardin DS, Rice J, Ahn C, Ferkol T, Howenstine M, Spears S, Prestidge C, Seilheimer DK, Shepherd R (March 2005). "Growth hormone treatment enhances nutrition and growth in children with cystic fibrosis receiving enteral nutrition". J. Pediatr. 146 (3): 324–8. doi:10.1016/j.jpeds.2004.10.037. PMID 15756212.
- ↑ Marks SC, Kissner DG (1997). "Management of sinusitis in adult cystic fibrosis". Am J Rhinol. 11 (1): 11–4. doi:10.2500/105065897781446810. PMID 9065342.
- ↑ Phillipson GT, Petrucco OM, Matthews CD (February 2000). "Congenital bilateral absence of the vas deferens, cystic fibrosis mutation analysis and intracytoplasmic sperm injection". Hum. Reprod. 15 (2): 431–5. doi:10.1093/humrep/15.2.431. PMID 10655317.
- ↑ Ciofu, O; Lykkesfeldt, J (Aug 7, 2014). "Antioxidant supplementation for lung disease in cystic fibrosis.". The Cochrane database of systematic reviews. 8: CD007020. doi:10.1002/14651858.CD007020.pub3. PMID 25102015.
- ↑ "What is the life expectancy for people who have CF (in the United States)?". Cystic Fibrosis Foundation. 2008. Retrieved 2010-03-14.
- ↑ MacKenzie, T; Gifford, AH; Sabadosa, KA; Quinton, HB; Knapp, EA; Goss, CH; Marshall, BC (Aug 19, 2014). "Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the cystic fibrosis foundation patient registry.". Annals of Internal Medicine. 161 (4): 233–41. doi:10.7326/m13-0636. PMID 25133359.
- ↑ "Canadian Cystic Fibrosis Patient Data Registry Report" (PDF). Canadian Cystic Fibrosis Foundation. 2007. Retrieved 2010-03-14.
- ↑ "Cystic Fibrosis Patient Registry Annual Data Report 2009" (PDF). Cystic Fibrosis Foundation. 2009.
- ↑ Yu H, Nasr SZ, Deretic V (April 2000). "Innate lung defenses and compromised Pseudomonas aeruginosa clearance in the malnourished mouse model of respiratory infections in cystic fibrosis". Infect. Immun. 68 (4): 2142–7. doi:10.1128/IAI.68.4.2142-2147.2000. PMC 97396. PMID 10722612.
- ↑ Ratjen F, Döring G (February 2003). "Cystic fibrosis". Lancet. 361 (9358): 681–9. doi:10.1016/S0140-6736(03)12567-6. PMID 12606185.
- ↑ Rosenstein BJ, Zeitlin PL (January 1998). "Cystic fibrosis". Lancet. 351 (9098): 277–82. doi:10.1016/S0140-6736(97)09174-5. PMID 9457113.
- ↑ Schmitz TG, Goldbeck L (2006). "The effect of inpatient rehabilitation programmes on quality of life in patients with cystic fibrosis: a multi-center study". Health Qual Life Outcomes. 4: 8. doi:10.1186/1477-7525-4-8. PMC 1373610. PMID 16457728.
- ↑ Hegarty M, Macdonald J, Watter P, Wilson C (July 2009). "Quality of life in young people with cystic fibrosis: effects of hospitalization, age and gender, and differences in parent/child perceptions". Child Care Health Dev. 35 (4): 462–8. doi:10.1111/j.1365-2214.2008.00900.x. PMID 18991968.
Havermans T, Vreys M, Proesmans M, De Boeck C (January 2006). "Assessment of agreement between parents and children on health-related quality of life in children with cystic fibrosis". Child Care Health Dev. 32 (1): 1–7. doi:10.1111/j.1365-2214.2006.00564.x. PMID 16398786. - ↑ Moorcroft AJ, Dodd ME, Webb AK (1998). "Exercise limitations and training for patients with cystic fibrosis". Disabil Rehabil. 20 (6–7): 247–53. doi:10.3109/09638289809166735. PMID 9637933.
- ↑ "Medications". Cystic Fibrosis Canada. 2011. No. 10684-5100 RR0001.
- ↑ Araújo FG, Novaes FC, Santos NP, Martins VC, Souza SM, Santos SE, Ribeiro-dos-Santos AK (January 2005). "Prevalence of deltaF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the North of Brazil". Braz. J. Med. Biol. Res. 38 (1): 11–5. doi:10.1590/S0100-879X2005000100003. PMID 15665983.
- ↑ Tobias, Edward (2011). Essential Medical Genetics. John Wiley & Sons. p. 312. ISBN 1-118-29370-3.
- ↑ "The Canadian Facts & Figures on Cystic Fibrosis".
- ↑ "Genetic Carrier Testing". Cystic Fibrosis Foundation. 2007.
- ↑ Rosenstein BJ, Cutting GR (April 1998). "The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel". J. Pediatr. 132 (4): 589–95. doi:10.1016/S0022-3476(98)70344-0. PMID 9580754.
- ↑ Hamosh A, FitzSimmons SC, Macek M, Knowles MR, Rosenstein BJ, Cutting GR (February 1998). "Comparison of the clinical manifestations of cystic fibrosis in black and white patients". J. Pediatr. 132 (2): 255–9. doi:10.1016/S0022-3476(98)70441-X. PMID 9506637.
- ↑ Farrell P, Joffe S, Foley L, Canny GJ, Mayne P, Rosenberg M (September 2007). "Diagnosis of cystic fibrosis in the Republic of Ireland: epidemiology and costs". Ir Med J. 100 (8): 557–60. PMID 17955689.
- ↑ Hytönen M, Patjas M, Vento SI, Kauppi P, Malmberg H, Ylikoski J, Kere J (December 2001). "Cystic fibrosis gene mutations deltaF508 and 394delTT in patients with chronic sinusitis in Finland". Acta Otolaryngol. 121 (8): 945–7. doi:10.1080/000164801317166835. PMID 11813900.
- ↑ "WHO | Genes and human disease". Who.int. 2010-12-07. Retrieved 2013-01-23.
- ↑ Russell, Peter (2011). Biology : the dynamic science. (2nd ed.). Belmont, CA: Brooks/Cole, Cengage Learning. p. 304. ISBN 978-0-538-49372-7.
- ↑ "Genetic testing for cystic fibrosis Genetic Testing for Cystic Fibrosis". Consensus Development Conference Statement. National Institutes of Health. April 14–16, 1997.
- ↑ Rosenfeld M, Davis R, FitzSimmons S, Pepe M, Ramsey B (May 1997). "Gender gap in cystic fibrosis mortality". Am. J. Epidemiol. 145 (9): 794–803. doi:10.1093/oxfordjournals.aje.a009172. PMID 9143209.
- ↑ Coakley RD, Sun H, Clunes LA, Rasmussen JE, Stackhouse JR, Okada SF, Fricks I, Young SL, Tarran R (December 2008). "17beta-Estradiol inhibits Ca2+
-dependent homeostasis of airway surface liquid volume in human cystic fibrosis airway epithelia". J. Clin. Invest. 118 (12): 4025–35. doi:10.1172/JCI33893. PMC 2582929. PMID 19033671. - ↑ Verma N, Bush A, Buchdahl R (October 2005). "Is there still a gender gap in cystic fibrosis?". Chest. 128 (4): 2824–34. doi:10.1378/chest.128.4.2824. PMID 16236961.
- ↑ Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W (September 2009). "Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality". Diabetes Care. 32 (9): 1626–31. doi:10.2337/dc09-0586. PMC 2732133. PMID 19542209.
- ↑ "CF worse for women 'due to effect of estrogen'". The Irish Times. August 8, 2010.
- ↑ Wennberg C, Kucinskas V (1994). "Low frequency of the delta F508 mutation in Finno-Ugrian and Baltic populations". Hum. Hered. 44 (3): 169–71. doi:10.1159/000154210. PMID 8039801.
- ↑ Kere J, Savilahti E, Norio R, Estivill X, de la Chapelle A (September 1990). "Cystic fibrosis mutation delta F508 in Finland: other mutations predominate". Hum. Genet. 85 (4): 413–5. doi:10.1007/BF02428286. PMID 2210753.
- ↑ Wiuf C (August 2001). "Do delta F508 heterozygotes have a selective advantage?". Genet. Res. 78 (1): 41–7. doi:10.1017/S0016672301005195. PMID 11556136.
- ↑ Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ (October 1994). "Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model". Science. 266 (5182): 107–9. Bibcode:1994Sci...266..107G. doi:10.1126/science.7524148. PMID 7524148.
- ↑ Cuthbert AW, Halstead J, Ratcliff R, Colledge WH, Evans MJ (January 1995). "The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study". J. Physiol. (Lond.). 482 (Pt 2): 449–54. doi:10.1113/jphysiol.1995.sp020531. PMC 1157742. PMID 7714835.
- ↑ Högenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, Rosenblatt RL, Prestidge CB, Fordtran JS (December 2000). "Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion". Am. J. Hum. Genet. 67 (6): 1422–7. doi:10.1086/316911. PMC 1287919. PMID 11055897.
- ↑ Pier GB, Grout M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, Ratcliff R, Evans MJ, Colledge WH (May 1998). "Salmonella typhi uses CFTR to enter intestinal epithelial cells". Nature. 393 (6680): 79–82. Bibcode:1998Natur.393...79P. doi:10.1038/30006. PMID 9590693.
- ↑ Modiano G, Ciminelli BM, Pignatti PF (March 2007). "Cystic fibrosis and lactase persistence: a possible correlation". Eur. J. Hum. Genet. 15 (3): 255–9. doi:10.1038/sj.ejhg.5201749. PMID 17180122.
- ↑ Poolman EM, Galvani AP (February 2007). "Evaluating candidate agents of selective pressure for cystic fibrosis". Journal of the Royal Society, Interface. 4 (12): 91–8. doi:10.1098/rsif.2006.0154. PMC 2358959. PMID 17015291.
- ↑ Williams, N (2006). "Footprint fears for new TB threat". Current Biology. 16 (19): R821. doi:10.1016/j.cub.2006.09.009.
- ↑ Tobacman, Joanne K. (2003-06-01). "Does deficiency of arylsulfatase B have a role in cystic fibrosis?". Chest. 123 (6): 2130–2139. doi:10.1378/chest.123.6.2130. ISSN 0012-3692. PMID 12796199.
- 1 2 3 Busch R (1990). "On the history of cystic fibrosis". Acta Univ Carol Med (Praha). 36 (1–4): 13–5. PMID 2130674.
- ↑ Fanconi, G.; Uehlinger, E.; Knauer, C. (1936). "Das coeliakiesyndrom bei angeborener zysticher pankreasfibromatose und bronchiektasien". Wien. Med. Wschr. 86: 753–6.
- ↑ Di Sant'Agnese PA, Darling RC, Perera GA, Shea E (November 1953). "Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease". Pediatrics. 12 (5): 549–63. PMID 13111855.
- ↑ Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL (September 1989). "Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA". Science. 245 (4922): 1066–73. Bibcode:1989Sci...245.1066R. doi:10.1126/science.2475911. PMID 2475911.
- ↑ Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N (September 1989). "Identification of the cystic fibrosis gene: chromosome walking and jumping". Science. 245 (4922): 1059–65. Bibcode:1989Sci...245.1059R. doi:10.1126/science.2772657. PMID 2772657.
- ↑ Lee, Tim WR (26 Nov 2013). "Topical cystic fibrosis transmembrane conductance regulator gene replacement for cystic fibrosis-related lung disease". Cochrane Database of Systematic Reviews. 11 (11): CD005599. doi:10.1002/14651858.CD005599.pub4. PMID 24282073. Retrieved 27 October 2014.
- ↑ Alton, EW; Armstrong, DK (3 July 2015). "Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial.". The Lancet. Respiratory medicine. 3: 684–91. doi:10.1016/S2213-2600(15)00245-3. PMID 26149841.
- ↑ Ramalho AS, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD (November 2002). "Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis". Am. J. Respir. Cell Mol. Biol. 27 (5): 619–27. doi:10.1165/rcmb.2001-0004oc. PMID 12397022.
- ↑ Tate S, Elborn S (March 2005). "Progress towards gene therapy for cystic fibrosis". Expert Opin Drug Deliv. 2 (2): 269–80. doi:10.1517/17425247.2.2.269. PMID 16296753.
- ↑ Online Mendelian Inheritance in Man (OMIM) CYSTIC FIBROSIS; CF -219700
- ↑ Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK, Nieuwenhuis EE, Beekman JM, Clevers H (Dec 5, 2013). "Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients". Cell Stem Cell. 13 (6): 653–8. doi:10.1016/j.stem.2013.11.002. PMID 24315439.
- ↑ Dietz HC (August 2010). "New therapeutic approaches to Mendelian disorders". N. Engl. J. Med. 363 (9): 852–63. doi:10.1056/NEJMra0907180. PMID 20818846. Free full text
- ↑ Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, Kerem B, Kerem E (October 2003). "Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations". N. Engl. J. Med. 349 (15): 1433–41. doi:10.1056/NEJMoa022170. PMID 14534336.
- ↑ Clinical trial number NCT00803205 for "Study of Ataluren (PTC124™) in Cystic Fibrosis" at ClinicalTrials.gov
- ↑ Cheng, K; Ashby, D; Smyth, RL (15 December 2014). "Ursodeoxycholic acid for cystic fibrosis-related liver disease.". The Cochrane database of systematic reviews. 12: CD000222. doi:10.1002/14651858.CD000222.pub3. PMID 25501301.
External links
| Wikimedia Commons has media related to Cystic fibrosis. |
- Cystic fibrosis at DMOZ
- cf at NIH/UW GeneTests
- CF-europe.eu
- Search GeneCards for genes involved in cystic fibrosis
- Cystic Fibrosis Mutation Database