X-inactivation


X-inactivation (also called lyonization) is a process by which one of the copies of the X chromosome present in female mammals is inactivated. The inactive X chromosome is silenced by its being packaged in such a way that it has a transcriptionally inactive structure called heterochromatin. As nearly all female mammals have two X chromosomes, X-inactivation prevents them from having twice as many X chromosome gene products as males, who only possess a single copy of the X chromosome (see dosage compensation). The choice of which X chromosome will be inactivated is random in placental mammals such as humans, but once an X chromosome is inactivated it will remain inactive throughout the lifetime of the cell and its descendants in the organism. Unlike the random X-inactivation in placental mammals, inactivation in marsupials applies exclusively to the paternally derived X chromosome.
History
In 1959 Susumu Ohno showed that the two X-chromosomes of mammals were different: one appeared similar to the autosomes; the other was condensed and heterochromatic.[3] This finding suggested, independently to two groups of investigators, that one of the X-chromosomes underwent inactivation. In 1961, Mary Lyon proposed the random inactivation of one female X chromosome to explain the mottled phenotype of female mice heterozygous for coat color genes.[4] The Lyon hypothesis also accounted for the findings that one copy of the X chromosome in female cells was highly condensed, and that mice with only one copy of the X chromosome developed as infertile females. This suggested[5] to Ernest Beutler, studying heterozygous females for Glucose-6-phosphate dehydrogenase (G6PD) deficiency, that there were two red cell populations of erythrocytes in such heterozygotes: deficient cells and normal cells,[6] depending on whether the inactivated X chromosome contains the normal or defective G6PD allele.
Mechanism
Timing
All mouse cells undergo an early, imprinted inactivation of the paternally-derived X chromosome in two-cell or four-cell stage embryos.[7][8][9] The extraembryonic tissues (which give rise to the placenta and other tissues supporting the embryo) retain this early imprinted inactivation, and thus only the maternal X chromosome is active in these tissues.
In the early blastocyst, this initial, imprinted X-inactivation is reversed in the cells of the inner cell mass (which give rise to the embryo), and in these cells both X chromosomes become active again. Each of these cells then independently and randomly inactivates one copy of the X chromosome.[9] This inactivation event is irreversible during the lifetime of the cell, so all the descendants of a cell which inactivated a particular X chromosome will also inactivate that same chromosome. This phenomenon, which can be observed in the coloration of tortoiseshell cats when females are heterozygous for the X-linked gene, should not be confused with mosaicism, which is a term that specifically refers to differences in the genotype of various cell populations in the same individual; X-inactivation, which is an epigenetic change that results in a different phenotype, is not a change at the genotypic level. For an individual cell or lineage the inactivation is therefore skewed or 'non-random', and this can give rise to mild symptoms in female 'carriers' of X-linked genetic disorders.[10]
X-inactivation is reversed in the female germline, so that all oocytes contain an active X chromosome.
Selection of one active X chromosome
Normal females possess two X chromosomes, and in any given cell one chromosome will be active (designated as Xa) and one will be inactive (Xi). However, studies of individuals with extra copies of the X chromosome show that in cells with more than two X chromosomes there is still only one Xa, and all the remaining X chromosomes are inactivated. This indicates that the default state of the X chromosome in females is inactivation, but one X chromosome is always selected to remain active.
It is understood that X-chromosome inactivation is a random process, occurring at about the time of gastrulation in the epiblast (cells that will give rise to the embryo). The maternal and paternal X chromosomes have an equal probability of inactivation. This would suggest that women would be expected to suffer from X-linked disorders approximately 50% as often as men (because women have two X chromosomes, while men have only one); however, in actuality, the occurrence of these disorders in females is much lower than that. One explanation for this disparity is that over 25% of genes on the inactivated X chromosome remain expressed, thus providing women with added protection against defective genes coded by the X-chromosome. Some suggest that this disparity must be proof of preferential (non-random) inactivation. Preferential inactivation of the paternal X-chromosome occurs in both marsupials and in cell lineages that form the membranes surrounding the embryo,[11] whereas in mammals either the maternally or the paternally derived X-chromosome may be inactivated in different cell lines.[12]
The time period for X-chromosome inactivation explains this disparity. Inactivation occurs in the epiblast during gastrulation, which gives rise to the embryo.[13] Inactivation occurs on a cellular level, resulting in a mosaic expression, in which patches of cells have an inactive maternal X-chromosome, while other patches have an inactive paternal X-chromosome. For example, a female heterozygous for haemophilia (an X-linked disease) would have about half of her liver cells functioning properly, which is typically enough to ensure normal blood clotting.[14][15] Chance could result in significantly more dysfunctional cells; however, such statistical extremes are unlikely. Genetic differences on the chromosome may also render one X-chromosome more likely to undergo inactivation. Also, if one X-chromosome has a mutation hindering its growth or rendering it non viable, cells which randomly inactivated that X will have a selective advantage over cells which randomly inactivated the normal allele. Thus, although inactivation is initially random, cells that inactivate a normal allele (leaving the mutated allele active) will eventually be overgrown and replaced by functionally normal cells in which nearly all have the same X-chromosome activated.[14]
It is hypothesized that there is an autosomally-encoded 'blocking factor' which binds to the X chromosome and prevents its inactivation. The model postulates that there is a limiting blocking factor, so once the available blocking factor molecule binds to one X chromosome the remaining X chromosome(s) are not protected from inactivation. This model is supported by the existence of a single Xa in cells with many X chromosomes and by the existence of two active X chromosomes in cell lines with twice the normal number of autosomes.[16]
Sequences at the X inactivation center (XIC), present on the X chromosome, control the silencing of the X chromosome. The hypothetical blocking factor is predicted to bind to sequences within the XIC.
Expression of X-Linked Disorders in Heterozygous Females
It’s easy to see the effect of female X heterozygosity in localized traits, such as the unique coat pattern of a calico cat. It can be more difficult, however, to fully understand the expression of un-localized traits in these females, such as the expression of disease.
Since males only have one X-chromosome, they will express all of the alleles on that chromosome. Females however, will primarily express the genes of the X-chromosome that remains active. In homozygous females, it doesn’t particularly matter which chromosome is inactivated, as the alleles are the same. However, in heterozygous females the inactivation of one chromosome over the other can have a direct impact on phenotype. Because of this phenomenon, there is an observed increase in phenotypic variation in heterozygous females than in homozygous.[17] There are many different ways in which the phenotypic variation can play out. In many cases, heterozygous females may be asymptomatic or only present minor symptoms of a given disorder, such as with X-linked adrenoleukodystrophy.[18]
The differentiation of phenotype in heterozygous females is furthered by the presence of X-inactivation skewing. Typically, each X-chromosome is silenced half of the time, this process is skewed when preferential inactivation of a chromosome occurs. It is thought that skewing happens either by chance or by a physical characteristic of a chromosome that may cause it to be silenced more or less often, such as an unfavorable mutation.[19][20]
On average, each X chromosome is inactivated half of the time, however 5-20% of "apparently normal" women display X-inactivation skewing.[19] In cases where skewing is present, a broad range of symptom expression can occur, resulting in expression varying from minor to severe depending on the skewing proportion. An extreme case of this was seen where monozygotic female twins had extreme variance in expression of Menkes disease (an X-linked disorder) resulting in the death of one twin while the other remained asymptomatic.[21]
It’s thought that X-inactivation skewing could be caused by issues in the mechanism that causes inactivation, or by issues in the chromosome itself.[19][20] However, the link between phenotype and skewing is still being questioned, and should be examined on a case-by-case basis. A study looking at both symptomatic and asymptomatic females who were heterozygous for Duchenne and Becker muscular dystrophies (DMD) found no apparent link between transcript expression and skewed X-Inactivation. The study suggests that both mechanisms are independently regulated, and there are other unknown factors at play.[22]
Chromosomal component
The X-inactivation center (or simply XIC) on the X chromosome is necessary and sufficient to cause X-inactivation. Chromosomal translocations which place the XIC on an autosome lead to inactivation of the autosome, and X chromosomes lacking the XIC are not inactivated.
The XIC contains four non-translated RNA genes, Xist, Tsix, Jpx and Ftx, which are involved in X-inactivation. The XIC also contains binding sites for both known and unknown regulatory proteins.
Xist and Tsix RNAs
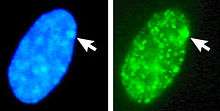
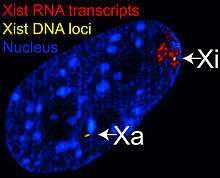
The X-inactive specific transcript (Xist) gene encodes a large non-coding RNA that is responsible for mediating the specific silencing of the X chromosome from which it is transcribed.[23] The inactive X chromosome is coated by Xist RNA,[24] whereas the Xa is not (See Figure to the right). The Xist gene is the only gene which is expressed from the Xi but not from the Xa. X chromosomes which lack the Xist gene cannot be inactivated.[25] Artificially placing and expressing the Xist gene on another chromosome leads to silencing of that chromosome.[26][27]
Prior to inactivation, both X chromosomes weakly express Xist RNA from the Xist gene. During the inactivation process, the future Xa ceases to express Xist, whereas the future Xi dramatically increases Xist RNA production. On the future Xi, the Xist RNA progressively coats the chromosome, spreading out from the XIC;[26] the Xist RNA does not localize to the Xa. The silencing of genes along the Xi occurs soon after coating by Xist RNA.
Like Xist, the Tsix gene encodes a large RNA which is not believed to encode a protein. The Tsix RNA is transcribed antisense to Xist, meaning that the Tsix gene overlaps the Xist gene and is transcribed on the opposite strand of DNA from the Xist gene.[28] Tsix is a negative regulator of Xist; X chromosomes lacking Tsix expression (and thus having high levels of Xist transcription) are inactivated much more frequently than normal chromosomes.
Like Xist, prior to inactivation, both X chromosomes weakly express Tsix RNA from the Tsix gene. Upon the onset of X-inactivation, the future Xi ceases to express Tsix RNA (and increases Xist expression), whereas Xa continues to express Tsix for several days.
Silencing
The inactive X chromosome does not express the majority of its genes, unlike the active X chromosome. This is due to the silencing of the Xi by repressive heterochromatin, which compacts the Xi DNA and prevents the expression of most genes.
Compared to the Xa, the Xi has high levels of DNA methylation, low levels of histone acetylation, low levels of histone H3 lysine-4 methylation, and high levels of histone H3 lysine-9 methylation and H3 lysine-27 methylation mark which is placed by the PRC2 complex recruited by Xist, all of which are associated with gene silencing.[29] Additionally, a histone variant called macroH2A (H2AFY) is exclusively found on nucleosomes along the Xi.[30][31]
Barr bodies
DNA packaged in heterochromatin, such as the Xi, is more condensed than DNA packaged in euchromatin, such as the Xa. The inactive X forms a discrete body within the nucleus called a Barr body.[32] The Barr body is generally located on the periphery of the nucleus, is late replicating within the cell cycle, and, as it contains the Xi, contains heterochromatin modifications and the Xist RNA.
Expressed genes on the inactive X chromosome
A fraction of the genes along the X chromosome escape inactivation on the Xi. The Xist gene is expressed at high levels on the Xi and is not expressed on the Xa.[33] Many other genes escape inactivation; some are expressed equally from the Xa and Xi, and others, while expressed from both chromosomes, are still predominantly expressed from the Xa.[34][35][36] Up to one quarter of genes on the human Xi are capable of escape.[34] Studies in the mouse suggest that in any given cell type, 3% to 15% of genes escape inactivation, and that escaping gene identity varies between tissues.[35][36]
Many of the genes which escape inactivation are present along regions of the X chromosome which, unlike the majority of the X chromosome, contain genes also present on the Y chromosome. These regions are termed pseudoautosomal regions, as individuals of either sex will receive two copies of every gene in these regions (like an autosome), unlike the majority of genes along the sex chromosomes. Since individuals of either sex will receive two copies of every gene in a pseudoautosomal region, no dosage compensation is needed for females, so it is postulated that these regions of DNA have evolved mechanisms to escape X-inactivation. The genes of pseudoautosomal regions of the Xi do not have the typical modifications of the Xi and have little Xist RNA bound.
The existence of genes along the inactive X which are not silenced explains the defects in humans with abnormal numbers of the X chromosome, such as Turner syndrome (X0) or Klinefelter syndrome (XXY). Theoretically, X-inactivation should eliminate the differences in gene dosage between affected individuals and individuals with a normal chromosome complement. In affected individuals, however, X-inactivation is incomplete and the dosage of these non-silenced genes will differ as they escape X-inactivation, similar to an autosomal aneuploidy.
The precise mechanisms that control escape from X-inactivation are not known, but silenced and escape regions have been shown to have distinct chromatin marks.[35][37] It has been suggested that escape from X-inactivation might be mediated by expression of long non-coding RNA (lncRNA) within the escaping chromosomal domains.[2]
Uses in experimental biology
Stanley Michael Gartler used X chromosome inactivation to demonstrate the clonal origin of cancers. Examining normal tissues and tumors from females heterozygous for isoenzymes of the sex-linked G6PD gene demonstrated that tumor cells from such individuals express only one form of G6PD, whereas normal tissues are composed of a nearly equal mixture of cells expressing the two different phenotypes. This pattern suggests that a single cell, and not a population, grows into a cancer.[38] However, this pattern has been proven wrong for many cancer types, suggesting that some cancers may be polyclonal in origin.[39]
Besides, measuring the methylation (inactivation) status of the polymorphic human androgen receptor (HUMARA) located on X-chromosome is considered the most accurate method to assess clonality in female cancer biopsies.[40] A great variety of tumors was tested by this method, some, such as renal cell carcinoma,[41] found monoclonal while others (e.g. mesothelioma[42]) were reported polyclonal.
Researchers have also investigated using X-chromosome inactivation to silence the activity of autosomal chromosomes. For example, Jiang et al. inserted a copy of the Xist gene into one copy of chromosome 21 in stem cells derived from an individual with trisomy 21 (Down's syndrome).[43] The inserted Xist gene induces Barr body formation, triggers stable heterochromatin modifications, and silences most of the genes on the extra copy of chromosome 21. In these modified stem cells, the Xist-mediated gene silencing seems to reverse some of the defects associated with Down's syndrome.
See also
- Sex-determination system
- Dosage compensation
- Barr body
- Heterochromatin
- Epigenetics
- Skewed X-inactivation
- Developmental disorders thought to be related to X-inactivation:
References
- ↑ Gartler SM, Varadarajan KR, Luo P, Canfield TK, Traynor J, Francke U, Hansen RS (2004). "Normal histone modifications on the inactive X chromosome in ICF and Rett syndrome cells: implications for methyl-/2/21/figure/F1 Figure 1". 2: 21. doi:10.1186/1741-7007-2-21.
- 1 2 Björn Reinius, Chengxi Shi, Liu Hengshuo, Kuljeet Singh Sandhu, Katarzyna J. Radomska, Glenn D. Rosen, Lu Lu, Klas Kullander, Robert W. Williams and Elena Jazin (November 2010). "Female-biased expression of long non-coding RNAs in domains that escape X-inactivation in mouse". BMC Genomics. 11: 614. doi:10.1186/1471-2164-11-614. PMC 3091755
 . PMID 21047393.
. PMID 21047393. - ↑ Ohno S, Kaplan WD, Kinosita R (1959). "Formation of the sex chromatin by a single X-chromosome in liver cells of rattus norvegicus". Exp Cell Res. 18 (2): 415–9. doi:10.1016/0014-4827(59)90031-X. PMID 14428474.
- ↑ Lyon MF (1961). "Gene Action in the X-chromosome of the Mouse (Mus musculus L.)". Nature. 190 (4773): 372–3. doi:10.1038/190372a0. PMID 13764598.
- ↑ Beutler, E (January 2008). "Glucose-6-phosphate dehydrogenase deficiency: a historical perspective". Blood. 111 (1): 16–24. doi:10.1182/blood-2007-04-077412. PMID 18156501.
- ↑ Beutler E, Yeh M, Fairbanks VF (January 1962). "The normal human female as a mosaic of X-chromosome activity: Studies using the genre for g-6-pd-deficiency as a marker". Proc. Natl. Acad. Sci. U.S.A. 48 (1): 9–16. doi:10.1073/pnas.48.1.9. PMC 285481. PMID 13868717.
- ↑ Takagi N, Sasaki M (1975). "Preferential inactivation of the paternally derived X chromosome in the extraembryonic membranes of the mouse". Nature. 256 (5519): 640–2. doi:10.1038/256640a0. PMID 1152998.
- ↑ Cheng MK, Disteche CM (2004). "Silence of the fathers: early X inactivation". BioEssays. 26 (8): 821–4. doi:10.1002/bies.20082. PMID 15273983.
- 1 2 Okamoto I, Otte A, Allis C, Reinberg D, Heard E (2004). "Epigenetic dynamics of imprinted X inactivation during early mouse development". Science. 303 (5658): 644–9. doi:10.1126/science.1092727. PMID 14671313.
- ↑ Puck, J; Willard, HF (1998). "X Inactivation in Females with X-Linked Disease". N. Engl. J. Med. 338 (5): 325–8. doi:10.1056/NEJM199801293380611. PMID 9445416.
- ↑ Graves JA (1996). "Mammals that break the rules: genetics of marsupials and monotremes". Annual Review of Genetics. 30: 233–260. doi:10.1146/annurev.genet.30.1.233. PMID 8982455.
- ↑ Lyon, Mary F. (1972-01-01). "X-Chromosome Inactivation and Developmental Patterns in Mammals". Biological Reviews. 47 (1): 1–35. doi:10.1111/j.1469-185X.1972.tb00969.x. PMID 4554151.
- ↑ Migeon, B (2010). "X chromosome inactivation in human cells". The Biomedical & Life Sciences Collection. http://hstalks.com/?t=BL0132567): Henry Stewart Talks, Ltd. pp. 1–54. Retrieved 15 December 2013.
- 1 2 Gartler, Stanley M; Goldman, Michael A (2001). "X-Chromosome Inactivation" (PDF). Encyclopedia of Life Sciences. www.els.net: Nature Publishing Group. pp. 1–2. Retrieved 10 November 2013.
- ↑ Connallon, Tim; Clark, Andrew G (2013). "Sex-Differential Selection and the Evolution of X Inactivation Strategies". Encyclopedia of Life Sciences. www.plosgenetics.org: PLOS Genetics. pp. 1–12. Retrieved 15 December 2013.
- ↑ Barakat, Tahsin Stefan. "X Chromosome Inactivation and Embryonic Stem Cells".
- ↑ Li, Ma (2015). "X-Inactivation Informs Variance-based Testing for X-linked Association of a Quantitative Trait". BMC Genomics.
- ↑ Habekost , Troller Clarissa, Santos Pereira Fernanda, Regla Vargas Carmen, Moura Coelho Daniella, Torrez Vitor, Pierre Oses Jean, Valmor Portela Luis, Schestatsky Pedro, Torres Felix Vitor, Matte Ursula, Leotti Torman Vanessa, Bannach Jardim Laura (2015). "Progression Rate of Myelopathy in X-linked Adrenoleukodystrophy Heterozygotes". Metabolic Brain Disease Metab Brain Dis. 30 (5): 1279–284. doi:10.1007/s11011-015-9672-2.
- 1 2 3 Belmont, John W. "Genetic Control of X Inactivation and Processes Leading to X-Inactivation Skewing." American Journal of Human Genetics 5.8 (1996): 1101-108. Web of Science. Web. 3 Mar. 2016.
- 1 2 Holle Jennifer R.; Marsh Rebecca A.; Maria Holdcroft Anna; Davies Stella M.; Wang Lijun; Zhang Kejian; Jordan Michael B. (2015). "Hemophagocytic Lymphohistiocytosis in a Female Patient Due to a Heterozygous XIAP Mutation and Skewed X Chromosome Inactivation". Pediatric Blood & Cancer Pediatr Blood Cancer. 62 (7): 1288–290. doi:10.1002/pbc.25483.
- ↑ Burgemeister , Lena Anna, Zirn Birgit, Oeffner Frank, Kaler Stephen G., Lemm Gunther, Rossier Eva, Büttel Hans-Martin (2015). "Menkes Disease with Discordant Phenotype in Female Monozygotic Twins". Am. J. Med. Genet. A. 167 (11): 2826–829. doi:10.1002/ajmg.a.37276.
- ↑ Brioschi, Simona, Francesca Gualandi, Chiara Scotton, Annarita Armaroli, Matteo Bovolenta, Maria S. Falzarano, Patrizia Sabatelli, Rita Selvatici, Adele D’Amico, Marika Pane, Giulia Ricci, Gabriele Siciliano, Silvana Tedeschi, Antonella Pini, Liliana Vercelli, Domenico De Grandis, Eugenio Mercuri, Enrico Bertini, Luciano Merlini, Tiziana Mongini, and Alessandra Ferlini. "Genetic Characterization in Symptomatic Female DMD Carriers: Lack of Relationship between X-inactivation, Transcriptional DMD Allele Balancing and Phenotype." BMC Medical Genetics BMC Med Genet 13.1 (2012): 73.
- ↑ Hoki Y, Kimura N, Kanbayashi M, Amakawa Y, Ohhata T, Sasaki H, Sado T (2009). "A proximal conserved repeat in the Xist gene is essential as a genomic element for X-inactivation in mouse". Development. 136 (1): 139–46. doi:10.1242/dev.026427. PMID 19036803.
- ↑ Ng K, Pullirsch D, Leeb M, Wutz A (2007). "Xist and the order of silencing" (Review Article). EMBO Rep. 8 (1): 34–9. doi:10.1038/sj.embor.7400871. PMC 1796754. PMID 17203100.
Figure 1 Xist RNA encompasses the X from which it is transcribed.
- ↑ Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N (1996). "Requirement for Xist in X chromosome inactivation". Nature. 379 (6561): 116–7. doi:10.1038/379131a0. PMID 8538762.
- 1 2 Herzing LB, Romer JT, Horn JM, Ashworth A (1997). "Xist has properties of the X-chromosome inactivation centre". Nature. 386 (6622): 272–5. doi:10.1038/386272a0. PMID 9069284.
- ↑ Lee JT, Jaenisch R (1997). "Long-range cis effects of ectopic X-inactivation centres on a mouse autosome". Nature. 386 (6622): 275–9. doi:10.1038/386275a0. PMID 9069285.
- ↑ Lee JT, Davidow LS, Warshawsky D (1999). "Tisx, a gene antisense to Xist at the X-inactivation centre". Nat Genet. 21 (4): 400–4. doi:10.1038/7734. PMID 10192391.
- ↑ Ng K, Pullirsch D, Leeb M, Wutz A (2007). "Xist and the order of silencing" (Review Article). EMBO Rep. 8 (1): 34–9. doi:10.1038/sj.embor.7400871. PMC 1796754. PMID 17203100.
Table 1 Features of the inactive X territory
– Originated from;
Chow J, Yen Z, Ziesche S, Brown C (2005). "Silencing of the mammalian X chromosome". Annu Rev Genomics Hum Genet. 6: 69–92. doi:10.1146/annurev.genom.6.080604.162350. PMID 16124854.
Lucchesi JC, Kelly WG, Panning B (2005). "Chromatin remodeling in dosage compensation". Annu. Rev. Genet. 39: 615–51. doi:10.1146/annurev.genet.39.073003.094210. PMID 16285873. - ↑ Costanzi C, Pehrson JR (1998). "Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals". Nature. 393 (6685): 599–601. doi:10.1038/31275. PMID 9634239.
- ↑ Costanzi C, Stein P, Worrad DM, Schultz RM, Pehrson JR (2000). "Histone macroH2A1 is concentrated in the inactive X chromosome of female preimplantation mouse embryos" (pdf). Development. 127 (11): 2283–9. PMID 10804171.
- ↑ Barr ML, Bertram EG (1949). "A Morphological Distinction between Neurones of the Male and Female, and the Behaviour of the Nucleolar Satellite during Accelerated Nucleoprotein Synthesis". Nature. 163 (4148): 676–7. doi:10.1038/163676a0. PMID 18120749.
- ↑ Plath K, Mlynarczyk-Evans S, Nusinow D, Panning B (2002). "Xist RNA and the mechanism of X chromosome inactivation". Annu Rev Genet. 36: 233–78. doi:10.1146/annurev.genet.36.042902.092433. PMID 12429693.
- 1 2 Carrel L, Willard H (2005). "X-inactivation profile reveals extensive variability in X-linked gene expression in females". Nature. 434 (7031): 400–404. doi:10.1038/nature03479. PMID 15772666.
- 1 2 3 Calabrese JM, Sun W, Song L, Mugford JW, Williams L, Yee D, Starmer J, Mieczkowski P, Crawford GE, Magnuson T (2012). "Site-specific silencing of regulatory elements as a mechanism of X inactivation". Cell. 151 (5): 951–63. doi:10.1016/j.cell.2012.10.037. PMC 3511858. PMID 23178118.
- 1 2 Yang F, Babak T, Shendure J, Disteche CM (2010). "Global survey of escape from X inactivation by RNA-sequencing in mouse". Genome Research. 20 (5): 614–22. doi:10.1101/gr.103200.109. PMC 2860163. PMID 20363980.
- ↑ Berletch JB, Yang F, Disteche CM (June 2010). "Escape from X inactivation in mice and humans". Genome Biology. 11 (6): 213. doi:10.1186/gb-2010-11-6-213. PMC 2911101. PMID 20573260.
- ↑ Linder D, Gartler SM (October 1965). "Glucose-6-phosphate dehydrogenase mosaicism: utilization as a cell marker in the study of leiomyomas". Science. 150 (3692): 67–9. doi:10.1126/science.150.3692.67. PMID 5833538.
- ↑ Parsons, BL (2008). "Many different tumor types have polyclonal tumor origin:evidence and implications". Mutat Res. 659 (3): 232–247. doi:10.1016/j.mrrev.2008.05.004. PMID 18614394.
- ↑ Chen, GL; Prchal, JT (2007). "X-linked clonality testing: interpretation and limitations". Blood. 110 (5): 1411–1419. doi:10.1182/blood-2006-09-018655. PMID 17435115.
- ↑ Petersson; et al. (2014). "The leiomyomatous stroma in renal cell carcinomas is polyclonal and not part of the neoplastic process". Virchows Arch. 465 (1): 89–96. doi:10.1007/s00428-014-1591-9. PMID 24838683.
- ↑ Comertpay; et al. (2014). "Evaluation of clonal origin of malignant mesothelioma". Journal of Translational Medicine. 12: 301. doi:10.1186/s12967-014-0301-3.
- ↑ Jiang; et al. (2013). "Translating dosage compensation to trisomy 21". Nature. 500 (7462): 296–300. doi:10.1038/nature12394. PMC 3848249. PMID 23863942.
- Huynh KD, Lee JT (2005). "X-chromosome inactivation: a hypothesis linking ontogeny and phylogeny". Nature Reviews Genetics. 6 (5): 410–18. doi:10.1038/nrg1604. PMID 15818384.
- X-inactivation as a possible cause for autoimmunity
Further reading
Review Article
- Goto T, Monk M (1 June 1998). "Regulation of X-Chromosome Inactivation in Development in Mice and Humans" (Review Article). Microbiol Mol Biol Rev. 62 (2): 362–78. PMC 98919. PMID 9618446.
- Lyon M (2003). "The Lyon and the LINE hypothesis". Semin Cell Dev Biol (Review Article). 14 (6): 313–8. doi:10.1016/j.semcdb.2003.09.015. PMID 15015738.
- Ng K, Pullirsch D, Leeb M, Wutz A (2007). "Xist and the order of silencing" (Review Article). EMBO Rep. 8 (1): 34–9. doi:10.1038/sj.embor.7400871. PMC 1796754. PMID 17203100.
- Cerase A, Pintacuda G, Tattermusch A, Avner P (2015). "Xist localization and function: new insights from multiple levels.". Genome Biology. 16: 166. doi:10.1186/s13059-015-0733-y. PMC 4539689. PMID 26282267.
| Wikimedia Commons has media related to X chromosome inactivation. |