Alpha-thalassemia
| Alpha-thalassemia | |
|---|---|
|
Alpha-thalassemia inheritance pattern | |
| Classification and external resources | |
| Specialty | hematology |
| ICD-10 | D56.0 |
| ICD-9-CM | 282.43 |
| OMIM | 141800 141850 142310 604131 |
| DiseasesDB | 448 33334 |
| eMedicine | article/955496 |
| MeSH | D017085 |
| GeneReviews | |
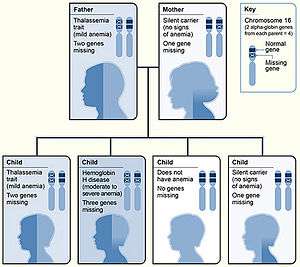
Alpha-thalassemia (α-thalassemia, α-thalassaemia) is a form of thalassemia involving the genes HBA1[1] and HBA2.[2] Alpha-thalassemia is due to impaired production of alpha chains from 1,2,3, or all 4 of the alpha globin genes, leading to a relative excess of beta globin chains. The degree of impairment is based on which clinical phenotype is present (how many genes are affected).[3][4]
Signs/symptoms
The presentation of individuals with alpha-thalassemia consist of the following:[4][5]
- Jaundice
- Fatigue
- Pronounced forehead
- Gallstones
- Hypertension (during pregnancy)
Cause
Alpha thalassemias are most commonly inherited in a Mendelian recessive manner. They are also associated with deletions of chromosome 16p.[6] Alpha thalassemia can also be acquired under rare circumstances.[7]
Pathophysiology
The mechanism sees that α thalassemias results in decreased alpha-globin production, therefore fewer alpha-globin chains are produced, resulting in an excess of beta chains in adults and excess γ chains in newborns. The excess β chains form unstable tetramers called Hemoglobin H or HbH of 4 beta chains. The excess γ chains form tetramers which are poor carriers of O2 since their affinity for O2 is too high so it is not dissociated in the periphery. Homozygote α0 thalassaemias, where there are lots of γ4 but no α-globins at all (referred to as Hb Barts), often result in death soon after birth.[3][5][8]
Diagnosis
Diagnosis of alpha thalassemia is primarily via laboratory evaluation and haemoglobin electrophoresis. Alpha-thalassemia can be mistaken for iron deficiency anaemia on a full blood count or blood film as both conditions have a microcytic anaemia. Serum iron and serum ferritin can be used to exclude iron deficiency anaemia.[3]
Types
There are two genetic loci for α globin, and thus four genes in diploid cells. Two genes are maternal in origin and two genes are paternal in origin. The severity of the α thalassemias is correlated with the number of affected α globin genes: the greater, the more severe will be the manifestations of the disease.When noting the genotype, a "α" indicates a functional alpha chain.[4][5][8]
| Alleles affected | Description | Genotype |
|---|---|---|
| One | This is known as alpha thalassemia silent and with this type there is minimal effect on hemoglobin synthesis. Three α-globin genes are enough to permit normal hemoglobin production, and there are no clinical symptoms. It occurs due to a deletion or non-deletion mutation.[5] | -/α α/α |
| Two |  Haematopoiesis(production of blood cells) Alpha thalassemia trait can exist in two forms:[5]
|
-/- α/α or -/α -/α |
| Three | This condition is called Hemoglobin H disease two unstable hemoglobins are present in the blood; Hemoglobin Barts (tetrameric γ chains) and Hemoglobin H (tetrameric β chains). Both of these unstable hemoglobins have a higher affinity for oxygen than normal hemoglobin.[9] There is a microcytic hypochromic anemia with target cells and Heinz bodies (precipitated HbH) on the peripheral blood smear, as well as hepatosplenomegaly. The disease is noticed in childhood or in early adult life anemia and hepatosplenomegaly are noted. | -/- -/α |
| Four | This is known as alpha thalassemia major, these fetuses are edematous and have little circulating hemoglobin, and the hemoglobin that is present is all tetrameric γ chains. When all four alleles are affected, the fetus likely will not survive gestation without in utero intervention; most infants with alpha thalassemia major are stillborn with hydrops fetalis. Fetuses treated with intrauterine transfusions throughout pregnancy starting at an early gestational age can survive to birth with acceptable morbidity. After birth, the treatment options include bone marrow transplantation or continued chronic transfusions.[10] | -/- -/- |
Treatment
Treatment for alpha thalassemia may consist of blood transfusions,and possible splenectomy, additionally gallstones may be a problem that would require surgery. Secondary complications from febrile episode should be monitored, having indicated this most individuals live without any need for treatment[5][11]
Additionally, stem cell transplantation should be considered as a treatment (and cure) which is best done in early age. Other options such as gene therapy are still being developed.[12]
Epidemiology
In terms of epidemiology, worldwide distribution of inherited alpha-thalassemia corresponds to areas of malaria exposure, suggesting a protective role. Thus, alpha-thalassemia is common in sub-Saharan Africa, the Mediterranean Basin, and generally tropical (and sub tropical) regions. The epidemiology of alpha-thalassemia in the US reflects this global distribution pattern. More specifically, HbH disease is seen in South East Asia, and the Middle East, while Hb Bart Hydrops fetalis is acknowledged in the South East Asia region only.[13]
See also
References
- ↑ Online Mendelian Inheritance in Man (OMIM) Hemoglobin—Alpha locus 1; HBA1 -141800
- ↑ Online Mendelian Inheritance in Man (OMIM) Hemoglobin—Alpha locus 2; HBA2 -141850
- 1 2 3 4 "Alpha Thalassemia Workup: Approach Considerations, Laboratory Studies, Hemoglobin Electrophoresis". emedicine.medscape.com. Retrieved 2016-05-24.
- 1 2 3 Reference, Genetics Home. "alpha thalassemia". Genetics Home Reference. Retrieved 8 September 2016.
- 1 2 3 4 5 6 7 Origa, Raffaella; Moi, Paolo; Galanello, Renzo; Cao, Antonio (1 January 1993). "Alpha-Thalassemia". GeneReviews(®). University of Washington, Seattle. Retrieved 22 September 2016.update 2013
- ↑ BRS Pathology (4th ed.). Lippincott Williams & Wilkins medical. December 2009. p. 162. ISBN 978-1451115871.
- ↑ Steensma DP, Gibbons RJ, Higgs DR (January 2005). "Acquired alpha-thalassemia in association with myelodysplastic syndrome and other hematologic malignancies". Blood. 105 (2): 443–52. doi:10.1182/blood-2004-07-2792. PMID 15358626.
- 1 2 Galanello, Renzo; Cao, Antonio (5 January 2011). "Alpha-thalassemia". Genetics in Medicine. 13 (2): 83–88. doi:10.1097/GIM.0b013e3181fcb468. ISSN 1098-3600. Retrieved 22 September 2016.
- ↑ "Hemoglobin H disease". Orphanet. Retrieved 22 September 2016.
- ↑ Vichinsky, Elliott P. (2009-01-01). "Alpha thalassemia major—new mutations, intrauterine management, and outcomes". ASH Education Program Book. 2009 (1): 35–41. doi:10.1182/asheducation-2009.1.35. ISSN 1520-4391. PMID 20008180.
- ↑ "Complications and Treatment | Thalassemia | Blood Disorders | NCBDDD | CDC". www.cdc.gov. Retrieved 22 September 2016.
- ↑ "Thalassaemia | Doctor | Patient". Patient. Retrieved 22 September 2016.
- ↑ Harteveld, Cornelis L; Higgs, Douglas R (2010). "α-thalassaemia". Orphanet Journal of Rare Diseases. 5 (1): 13. doi:10.1186/1750-1172-5-13. ISSN 1750-1172. PMC 2887799
 .
.
Further reading
- Anie, Kofi A; Massaglia, Pia (6 March 2014). "Psychological therapies for thalassaemia". Cochrane Database of Systematic Reviews. John Wiley & Sons, Ltd. doi:10.1002/14651858.cd002890.pub2/full. Retrieved 15 September 2016.
- Galanello, Renzo; Cao, Antonio (5 January 2011). "Alpha-thalassemia". Genetics in Medicine. 13 (2): 83–88. doi:10.1097/GIM.0b013e3181fcb468. ISSN 1098-3600. Retrieved 15 September 2016.
External links
- "What Are Thalassemias? - NHLBI, NIH". www.nhlbi.nih.gov. Retrieved 15 September 2016.