Indole
 | |
 | |
 | |
| Names | |
|---|---|
| IUPAC name
Indole | |
| Other names
2,3-Benzopyrrole, ketole, 1-benzazole | |
| Identifiers | |
| 120-72-9 | |
| 3D model (Jmol) | Interactive image |
| ChEBI | CHEBI:16881 |
| ChEMBL | ChEMBL15844 |
| ChemSpider | 776 |
| ECHA InfoCard | 100.004.019 |
| KEGG | C00463 |
| PubChem | 798 |
| RTECS number | NL2450000 |
| UNII | 8724FJW4M5 |
| |
| |
| Properties | |
| C8H7N | |
| Molar mass | 117.15 g/mol |
| Appearance | White solid |
| Odor | Feces or jasmine like |
| Density | 1.1747 g/cm3, solid |
| Melting point | 52 to 54 °C (126 to 129 °F; 325 to 327 K) |
| Boiling point | 253 to 254 °C (487 to 489 °F; 526 to 527 K) |
| 0.19 g/100 ml (20 °C) Soluble in hot water | |
| Acidity (pKa) | 16.2 (21.0 in DMSO) |
| Basicity (pKb) | 17.6 |
| Structure | |
| Pna21 | |
| Planar | |
| 2.11 D in benzene | |
| Hazards | |
| Safety data sheet | |
| R/S statement | R: 21/22-37/38-41-50/53 S: 26-36/37/39-60-61 |
| Flash point | 121 °C (250 °F; 394 K) |
| Related compounds | |
| Related aromatic compounds |
benzene, benzofuran, carbazole, carboline, indene, indoline, isatin, methylindole, oxindole, pyrrole, skatole |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| | |
| Infobox references | |
Indole is an aromatic heterocyclic organic compound with formula C8H7N. It has a bicyclic structure, consisting of a six-membered benzene ring fused to a five-membered nitrogen-containing pyrrole ring. Indole is widely distributed in the natural environment and can be produced by a variety of bacteria. As an intercellular signal molecule, indole regulates various aspects of bacterial physiology, including spore formation, plasmid stability, resistance to drugs, biofilm formation, and virulence.[1] The amino acid tryptophan is an indole derivative and the precursor of the neurotransmitter serotonin.[2]
General properties and occurrence
Indole is a solid at room temperature. Indole can be produced by bacteria as a degradation product of the amino acid tryptophan. It occurs naturally in human feces and has an intense fecal odor. At very low concentrations, however, it has a flowery smell,[3] and is a constituent of many flower scents (such as orange blossoms) and perfumes. It also occurs in coal tar.
The corresponding substituent is called indolyl.
Indole undergoes electrophilic substitution, mainly at position 3 (see diagram in right margin). Substituted indoles are structural elements of (and for some compounds, the synthetic precursors for) the tryptophan-derived tryptamine alkaloids like the neurotransmitter serotonin, and melatonin. Other indolic compounds include the plant hormone auxin (indolyl-3-acetic acid, IAA), tryptophol, the anti-inflammatory drug indomethacin, the betablocker pindolol, and the naturally occurring hallucinogen dimethyltryptamine.
The name indole is a portmanteau of the words indigo and oleum, since indole was first isolated by treatment of the indigo dye with oleum.
History

Indole chemistry began to develop with the study of the dye indigo. Indigo can be converted to isatin and then to oxindole. Then, in 1866, Adolf von Baeyer reduced oxindole to indole using zinc dust.[4] In 1869, he proposed a formula for indole (left).[5]
Certain indole derivatives were important dyestuffs until the end of the 19th century. In the 1930s, interest in indole intensified when it became known that the indole substituent is present in many important alkaloids (e.g., tryptophan and auxins), and it remains an active area of research today.[6]
Occurrence in nature
Indole is biosynthesized via anthranilate.[2] It condenses with serine by Michael addition of indole to PLP-aminoacrylate.
.svg.png) Indole is produced via anthranilate and is alkylated to give the amino acid tryptophan.
Indole is produced via anthranilate and is alkylated to give the amino acid tryptophan.
Indole is a major constituent of coal tar, and the 220–260 °C distillation fraction is the main industrial source of the material.
Synthetic routes
Indole and its derivatives can also be synthesized by a variety of methods.[7][8][9]
The main industrial routes start from aniline via vapor-phase reaction with ethylene glycol in the presence of catalysts:
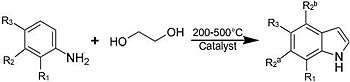
In general, reactions are conducted between 200 and 500 °C. Yields can be as high as 60%. Other precursors to indole include formyltoluidine, 2-ethylaniline, and 2-(2-nitrophenyl)ethanol, all of which undergo cyclizations.[10] Many other methods have been developed.
Leimgruber–Batcho indole synthesis
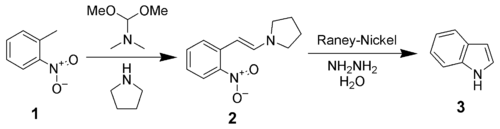
The Leimgruber–Batcho indole synthesis is an efficient method of synthesizing indole and substituted indoles. Originally disclosed in a patent in 1976, this method is high-yielding and can generate substituted indoles. This method is especially popular in the pharmaceutical industry, where many pharmaceutical drugs are made up of specifically substituted indoles.
Fischer indole synthesis
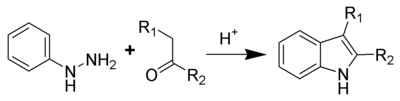

One of the oldest and most reliable methods for synthesizing substituted indoles is the Fischer indole synthesis, developed in 1883 by Emil Fischer. Although the synthesis of indole itself is problematic using the Fischer indole synthesis, it is often used to generate indoles substituted in the 2- and/or 3-positions. Indole can still be synthesized, however, using the Fischer indole synthesis by reacting phenylhydrazine with pyruvic acid followed by decarboxylation of the formed indole-2-carboxylic acid. This has also been accomplished in a one-pot synthesis using microwave irradiation.[11]
Other indole-forming reactions
- Bartoli indole synthesis
- Bischler–Möhlau indole synthesis
- Fukuyama indole synthesis
- Gassman indole synthesis
- Hemetsberger indole synthesis
- Larock indole synthesis
- Madelung synthesis
- Nenitzescu indole synthesis
- Reissert indole synthesis
- Baeyer–Emmerling indole synthesis
- In the Diels–Reese reaction [12][13] dimethyl acetylenedicarboxylate reacts with diphenylhydrazine to an adduct, which in xylene gives dimethyl indole-2,3-dicarboxylate and aniline. With other solvents, other products are formed: with glacial acetic acid a pyrazolone, and with pyridine a quinoline.
Chemical reactions of indole
Basicity
Unlike most amines, indole is not basic. The bonding situation is analogous to that in pyrrole. Strong acids such as hydrochloric acid can protonate indole. Indole is primarily protonated at the C3, rather than N1, owing to the enamine-like reactivity of the portion of the molecule located outside of the benzene ring. The protonated form has an pKa of −3.6. The sensitivity of many indolic compounds (e.g., tryptamines) under acidic conditions is caused by this protonation.
Electrophilic substitution
The most reactive position on indole for electrophilic aromatic substitution is C3, which is 1013 times more reactive than benzene. For example, it is alkylated by phosphorylated serine in the biosynthesis of the amino acid tryptophan (see figure above). Vilsmeier–Haack formylation of indole[14] will take place at room temperature exclusively at C3.
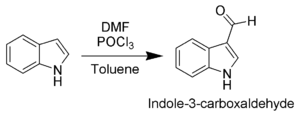
Since the pyrrollic ring is the most reactive portion of indole, electrophilic substitution of the carbocyclic (benzene) ring generally takes place only after N1, C2, and C3 are substituted. A noteworthy exception occurs when electrophilic substitution is carried out in conditions sufficiently acidic to exhaustively protonate C3. In this case, C5 is the most common site of electrophilic attack.[15]
Gramine, a useful synthetic intermediate, is produced via a Mannich reaction of indole with dimethylamine and formaldehyde. It is the precursor to indole-3-acetic acid and synthetic tryptophan.
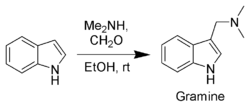
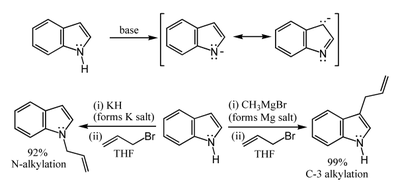
N–H acidity and organometallic indole anion complexes
The N–H center has a pKa of 21 in DMSO, so that very strong bases such as sodium hydride or n-butyl lithium and water-free conditions are required for complete deprotonation. The resulting organometalic derivatives can react in two ways. The more ionic salts such as the sodium or potassium compounds tend to react with electrophiles at nitrogen-1, whereas the more covalent magnesium compounds (indole Grignard reagents) and (especially) zinc complexes tend to react at carbon 3 (see figure below). In analogous fashion, polar aprotic solvents such as DMF and DMSO tend to favour attack at the nitrogen, whereas nonpolar solvents such as toluene favour C3 attack.[16]
Carbon acidity and C2 lithiation
After the N–H proton, the hydrogen at C2 is the next most acidic proton on indole. Reaction of N-protected indoles with butyl lithium or lithium diisopropylamide results in lithiation exclusively at the C2 position. This strong nucleophile can then be used as such with other electrophiles.
Bergman and Venemalm developed a technique for lithiating the 2-position of unsubstituted indole.[17]
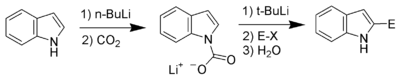
Alan Katritzky also developed a technique for lithiating the 2-position of unsubstituted indole.[18]
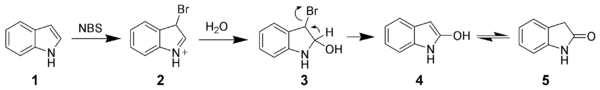
Oxidation of indole
Due to the electron-rich nature of indole, it is easily oxidized. Simple oxidants such as N-bromosuccinimide will selectively oxidize indole 1 to oxindole (4 and 5).
Cycloadditions of indole
Only the C2–C3 pi bond of indole is capable of cycloaddition reactions. Intramolecular variants are often higher-yielding than intermolecular cycloadditions. For example, Padwa et al.[19] have developed this Diels-Alder reaction to form advanced strychnine intermediates. In this case, the 2-aminofuran is the diene, whereas the indole is the dienophile. Indoles also undergo intramolecular [2+3] and [2+2] cycloadditions.

Despite mediocre yields, intermolecular cycloadditions of indole derivatives have been well documented.[20][21][22][23] One example is the Pictet-Spengler reaction between tryptophan derivatives and aldehydes.[24] The Pictet-Spengler reaction of indole derivatives, such as tryptophan, leads to a mixture of diastereomers as products. The formation of multiple products reduces the chemical yield of the desired product.
Applications
Natural jasmine oil, used in the perfume industry, contains around 2.5% of indole. Since 1 kilogram of the natural oil requires processing several million jasmine blossoms and costs around US$10,000, indole (among other things) is used in the manufacture of synthetic jasmine oil, costing about US$10 per kilogram.
See also
- Indole-3-butyric acid
- Indole test
- Isoindole
- Isoindoline
- Martinet dioxindole synthesis
- Skatole (3-methylindole)
- Stollé synthesis
- Tryptamine
References
- ↑ Lee, Jin-Hyung; Lee, Jintae (2010). "Indole as an intercellular signal in microbial communities". FEMS Microbiology Reviews. doi:10.1111/j.1574-6976.2009.00204.x. ISSN 0168-6445.
- 1 2 Nelson, David L.; Cox, Michael M. (2005), Principles of Biochemistry (4th ed.), New York: W. H. Freeman, ISBN 0-7167-4339-6
- ↑ http://www.leffingwell.com/olfact5.htm
- ↑ Baeyer, A. (1866). "Ueber die Reduction aromatischer Verbindungen mittelst Zinkstaub" [On the reduction of aromatic compounds by means of zinc dust]. Annalen der Chemie und Pharmacie. 140 (3): 295–296. doi:10.1002/jlac.18661400306.
- ↑ Baeyer, A.; Emmerling, A. (1869). "Synthese des Indols" [Synthesis of indole]. Berichte der Deutschen Chemischen Gesellschaft. 2: 679–682. doi:10.1002/cber.186900201268.
- ↑ Van Order, R. B.; Lindwall, H. G. (1942). "Indole". Chem. Rev. 30: 69–96. doi:10.1021/cr60095a004.
- ↑ Gribble, G. W. (2000). "Recent developments in indole ring synthesis—methodology and applications". J. Chem. Soc. Perkin Trans. 1 (7): 1045. doi:10.1039/a909834h.
- ↑ Cacchi, S.; Fabrizi, G. (2005). "Synthesis and Functionalization of Indoles Through Palladium-catalyzed Reactions". Chem. Rev. 105 (7): 2873–2920. doi:10.1021/cr040639b. PMID 16011327.
- ↑ Humphrey, G. R.; Kuethe, J. T. (2006). "Practical Methodologies for the Synthesis of Indoles". Chem. Rev. 106 (7): 2875–2911. doi:10.1021/cr0505270. PMID 16836303.
- ↑ Collin, Gerd; Höke, Hartmut (2005), "Indole", Ullmann's Encyclopedia of Industrial Chemistry, Weinheim: Wiley-VCH, doi:10.1002/14356007.a14_167
- ↑ Bratulescu, George (2008). "A new and efficient one-pot synthesis of indoles". Tetrahedron Letters. 49 (6): 984. doi:10.1016/j.tetlet.2007.12.015.
- ↑ Diels, Otto; Reese, Johannes (1934). "Synthesen in der hydroaromatischen Reihe. XX. Über die Anlagerung von Acetylen-dicarbonsäureester an Hydrazobenzol" [Syntheses in the hydroaromatic series. XX. The addition of acetylene dicarboxylic acid ester to hydrazobenzene]. Ann. 511: 168. doi:10.1002/jlac.19345110114.
- ↑ Huntress, Ernest H.; Bornstein, Joseph; Hearon, William M. (1956). "An Extension of the Diels-Reese Reaction". J. Am. Chem. Soc. 78 (10): 2225. doi:10.1021/ja01591a055.
- ↑ James, P. N.; Snyder, H. R. (1959). "Indole-3-aldehyde". Organic Syntheses. 39: 30. doi:10.15227/orgsyn.039.0030.
- ↑ Noland, W. E.; Rush, K. R.; Smith, L. R. (1966). "Nitration of Indoles. IV. The Nitration of 2-Phenylindole.". J. Org. Chem. 31: 65–69. doi:10.1021/jo01339a013.
- ↑ Heaney, H.; Ley, S. V. (1974). "1-Benzylindole". Organic Syntheses. 54: 58. doi:10.15227/orgsyn.054.0058.
- ↑ Bergman, J.; Venemalm, L. (1992). "Efficient synthesis of 2-chloro-, 2-bromo-, and 2-iodoindole". J. Org. Chem. 57 (8): 2495. doi:10.1021/jo00034a058.
- ↑ Katritzky, Alan R.; Li, Jianqing; Stevens, Christian V. (1995). "Facile Synthesis of 2-Substituted Indoles and Indolo[3,2-b]carbazoles from 2-(Benzotriazol-1-ylmethyl)indole". J. Org. Chem. 60 (11): 3401–3404. doi:10.1021/jo00116a026.
- ↑ Lynch, S. M.; Bur, S. K.; Padwa, A. (2002). "Intramolecular Amidofuran Cycloadditions across an Indole π-Bond: An Efficient Approach to the Aspidosperma and Strychnos ABCE Core". Org. Lett. 4 (26): 4643–5. doi:10.1021/ol027024q. PMID 12489950.
- ↑ Cox, E. D.; Cook, J. M. (1995). "The Pictet-Spengler condensation: a new direction for an old reaction". Chemical Reviews. 95 (6): 1797–1842. doi:10.1021/cr00038a004.
- ↑ Gremmen, C.; Willemse, B.; Wanner, M. J.; Koomen, G.-J. (2000). "Enantiopure Tetrahydro-β-carbolines via Pictet–Spengler Reactions with N-Sulfinyl Tryptamines". Org. Lett. 2 (13): 1955–1958. doi:10.1021/ol006034t.
- ↑ Larghi, Enrique L.; Amongero, Marcela; Bracca, Andrea B. J.; Kaufman, Teodoro S. (2005). "The intermolecular Pictet–Spengler condensation with chiral carbonyl derivatives in the stereoselective syntheses of optically-active isoquinoline and indole alkaloids". Arkivoc. RL-1554K: 98–153.
- ↑ Kaufman, Teodoro S. (2005). "Synthesis of Optically-Active Isoquinoline and Indole Alkaloids Employing the Pictet–Spengler Condensation with Removable Chiral Auxiliaries Bound to Nitrogen". In Vicario, J. L. New Methods for the Asymmetric Synthesis of Nitrogen Heterocycles. Thiruvananthapuram: Research SignPost. p. 99–147. ISBN 81-7736-278-X.
- ↑ Bonnet, D.; Ganesan, A. (2002). "Solid-Phase Synthesis of Tetrahydro-β-carbolinehydantoins via the N-Acyliminium Pictet–Spengler Reaction and Cyclative Cleavage". J. Comb. Chem. 4 (6): 546–548. doi:10.1021/cc020026h.
General references
- Houlihan, W. J., ed. (1972). Indoles Part One. New York: Wiley Interscience.
- Sundberg, R. J. (1996). Indoles. San Diego: Academic Press. ISBN 0-12-676945-1.
- Joule, J. A.; Mills, K. (2000). Heterocyclic Chemistry. Oxford, UK: Blackwell Science. ISBN 0-632-05453-0.
- Joule, J. (2000). E. J., Thomas, ed. Science of Synthesis. 10. Stuttgart: Thieme. p. 361. ISBN 3-13-112241-2.
- Schoenherr, H.; Leighton, J. L. (2012). Direct and Highly Enantioselective Iso-Pictet-Spengler Reactions with α-Ketoamides: Access to Underexplored Indole Core Structures. Org. Lett. 14. p. 2610.
External links
| Wikimedia Commons has media related to Indoles. |
- Synthesis of indoles (overview of recent methods)
- Synthesis and propierties of indoles at chemsynthesis.com