Epigenetics of neurodegenerative diseases
.gif)
Neurodegenerative diseases are a heterogenous group of complex disorders linked by the degeneration of neurons in either the peripheral nervous system or the central nervous system. Their underlying causes are extremely variable and complicated by various genetic and/or environmental factors. These diseases cause progressive deterioration of the neuron resulting in decreased signal transduction and in some cases even neuronal death. Peripheral nervous system (PNS) diseases may be further categorized by the type of nerve cell (motor, sensory, or both) affected by the disorder. Effective treatment of these diseases is often prevented by lack of understanding of the underlying molecular and genetic pathology. Epigenetic therapy is being investigated as a method of correcting the expression levels of misregulated genes in neurodegenerative diseases.
Neurodengenerative diseases of motor neurons can cause degeneration of motor neurons involved in voluntary muscle control such as muscle contraction and relaxation. This article will cover the epigenetics and treatment of amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA). See the Motor Neuron Fact Sheet for details regarding other motor neuron diseases.Neurodegenerative diseases of the central nervous system can affect the brain and/or spinal cord. This article will cover the epigenetics and treatment of Alzheimer’s disease (AD), Huntington’s disease (HD), and Parkinson’s disease (PD). These diseases are characterized by chronic and progressive neuronal dysfunction, sometimes leading to behavioral abnormalities (as with PD), and, ultimately, neuronal death, resulting in dementia.
Neurodegeneration Neurodegenerative diseases of sensory neurons can cause degeneration of sensory neurons involved in transmitting sensory information such as hearing and seeing. The main group of sensory neuron diseases are hereditary sensory and autonomic neuropathies (HSAN) such as HSAN I, HSAN II, and Charcot-Marie-Tooth Type 2B (CMT2B).[1] Though some sensory neuron diseases are recognized as neurodegenerative, epigenetic factors have not yet been clarified in the molecular pathology and as such will not be covered in this article.
Epigenetics and epigenetic drugs
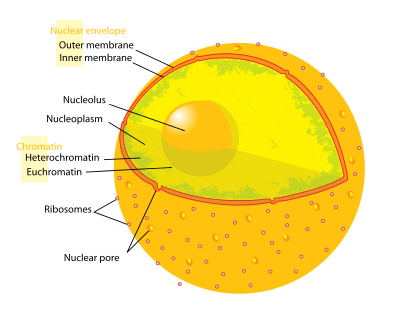
The term epigenetics refers to three levels of gene regulation: (1) DNA methylation, (2) histone modifications, and (3) non-coding RNA (ncRNA) function. Briefly, histone-mediated transcriptional control occurs by the wrapping of DNA around a histone core. This DNA-histone structure is called a nucleosome; the more tightly the DNA is bound by the nucleosome, and the more tightly a string of nucleosomes are compressed among each other, the greater the repressive effect on transcription of genes in the DNA sequences near or wrapped around the histones, and vice versa (i.e. looser DNA binding and relaxed compaction leads to a comparatively derepressed state, resulting in facultative heterochromatin or, even further derepressed, euchromatin). At its most repressive state, involving many folds into itself and other scaffolding proteins, DNA-histone structures form constitutive heterochromatin. This chromatin structure is mediated by these three levels of gene regulation. The most relevant epigenetic modifications to treatment of neurodegenerative diseases are DNA methylation and histone protein modifications via methylation or acetylation.[2]
- In mammals, methylation occurs on DNA and histone proteins. DNA methylation occurs on the cytosine of CpG dinucleotides in the genomic sequence, and protein methylation occurs on the amino termini of the core histone proteins - most commonly on lysine residues (Bradley et al., 2007). CpG refers to a dinucleotide composed of a cytosine deoxynucleotide immediately adjacent to a guanine deoxynucleotide. A cluster of CpG dinucleotides clustered together is called a CpG island, and in mammals, these CpG islands are one of the major classes of gene promoters, onto or around which transcription factors may bind and transcription can begin. Methylation of CpG dinucleotides and/or islands within gene promoters is associated with transcriptional repression via interference of transcription factor binding and recruitment of transcriptional repressors with methyl binding domains. Methylation of intragenic regions is associated with increased transcription. The group of enzymes responsible for addition of methyl groups to DNA are called DNA methyltransferases (DNMTs). The enzyme responsible for removal of methyl group are called DNA demethylases. The effects of histone methylation are residue dependent (e.g. which amino acid on which histone tail is methylated) therefore the resulting transcriptional activity and chromatin regulation can vary.[3] The enzymes responsible for the addition of methyl groups to histones are called histone methyltransferases (HMTs). The enzymes responsible for the removal of methyl groups from histone are histone demethylases.
- Acetylation occurs on the lysine residues found at the amino N-terminal of histone tails. Histone acetylation is most commonly associated with relaxed chromatin, transcriptional derepression, and thus actively transcribed genes (Bradley et al., 2007). Histone acetyltransferases (HATs) are enzymes responsible for the addition of acetyl groups, and histone deacetylases (HDACs) are enzymes responsible for the removal of acetyl groups. Therefore, the addition or removal of an acetyl group to a histone can alter the expression of nearby genes. The majority of drugs being investigated are inhibitors of proteins that remove acetyl from histones or histone deacetylases (HDACs).
- Briefly, ncRNAs are involved in signaling cascades with epigenetic marking enzymes such as HMTs, and/or with RNA interference(RNAi) machinery. Frequently these signaling cascades result in epigenetic repression (for one example, see X-chromosome inactivation), though there are some cases in which the opposite is true. For example, BACE1-AS ncRNA expression is upregulated in Alzheimer's disease patients and results in increased stability of BACE1 - the mRNA precursor to an enzyme involved in Alzheimer's disease (Faghihi et al., 2008). See below for more detail.
Epigenetic drugs target the proteins responsible for modifications on DNA or histone. Current epigenetic drugs include but are not limited to: HDAC inhibitors (HDACi), HAT modulators, DNA methyltransferase inhibitors, and histone demethylase inhibitors (Urdinguio et al., 2009; Peedicayil, 2013). The majority of epigenetic drugs tested for use against neurodegenerative diseases are HDAC inhibitors; however, some DNMT inhibitors have been tested as well. While the majority of epigenetic drug treatments have been conducted in mouse models, some experiments have been performed on human cells as well as in human drug trials (see table below). There are inherent risks in using epigenetic drugs as therapies for neurodegenerative disorders as some epigenetic drugs (e.g. HDACis such as sodium butyrate) are non-specific in their targets, which leaves potential for off-target epigenetic marks causing unwanted epigenetic modifications.
| Function | Classification | Drug | ALS | AD | HD | PD | SMA |
|---|---|---|---|---|---|---|---|
| DNA-methylation inhibitor | chemical analogue of cytidine | Azathioprine | M (ny) | M (ny) | |||
| HDAC inhibitor (small molecule) | benzamide | M344 | MC 19 | ||||
| fatty acid | Sodium butyrate | M (y) 5, 6, 7 ; H (ny) | D (y) 11 | M (y) 14; R (y) 15;
D (y) 16, 18; H (ny) |
MC 20; M (y) 21; H (ny) | ||
| Sodium phenylbutyrate | M (y) 1; H (y) 2 | M (y) 8; H (ny) | H (ys) 12 | MC 20; H (v) 21, 22 | |||
| Valproic acid | M (y) 2; H (ni) 3 | M (y) 9; H (ny) | D (y) 11 | R (y) 17; H (ny) | MC 23, 24; M (y) 25;
H (v) 26, 27, 28, 29 | ||
| hydroxamic acid | Trichostatin A | M (y) 4; H (ny) | M (y) 10; H (ny) | MC 13; D (y) 11 | M (y) 30, 31; H (ny) | ||
| Vorinostat (suberanilohydroxamic acid-SAHA) | M (y) 9; H (ny) | MC 13; D (y) 11 | D (y) 18 | MC 32, 33; M (y) 34; H (ny) |
- Disease: amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Huntington's disease (HD), spinal muscular atrophy (SMA), Parkinson's disease (PD)
- Tested on: mouse (M), only mouse cells (MC), human (H), Drosophila (D), rat (R)
- Successful treatment: yes (y), yes but with side effects (ys), not yet (ny), variable (v), no improvement (ni)
- References: listed by column (disease) and by ascending row (drug) order
- ALS: (1) Del Signore et al., 2009; Petri et al., 2006 (2) Cudkowicz et al., 2009 (3) Piepers et al., 2009; (4) Yoo et al., 2011
- AD: (5) Fischer et al., 2007; (6) Ricobaraza et al., 2010; (7) Govindarajan et al., 2011; (8) Ricobaraza et al., 2012; (9) Kilgore et al., 2010; (10) Francis et al., 2009
- HD: (11) Steffan et al., 2001 (12) Gardian et al., 2005 (13) Dompierre et al., 2007
- PD: (14) Zhou et al., 2011 (15) Rane et al., 2012 (16) St. Laurent et al., 2013 (17) Monti et al., 2010 (18) Kontopolous et al., 2006
- SMA: (19) Riessland et al., 2006 (20) Andreassi et al., 2004 (21) Mercuri et al., 2007 (22) Brahe et al., 2005 (23) Sumner et al., 2003 (24) Brichta et al., 2003 (25) Tsai et al., 2006 (26) Weihl et al., 2006 (27) Piepers et al., 2010 (28) Swoboda et al., 2010 (29) Darbar et al., 2011 (30) Narver et al., 2008 (31) Avila et al., 2007 (32) Hahnan et al., 2006 (33) Kernochan et al., 2005 (34) Riessland et al., 2010
Neurodegenerative diseases of motor neurons
Amyotrophic lateral sclerosis (ALS)
Amyotrophic Lateral Sclerosis (ALS), also known as Lou Gehrig’s disease, is a motor neuron disease that involves neurogeneration. All skeletal muscles in the body are controlled by motor neurons that communicate signals from the brain to the muscle through a neuromuscular junction. When the motor neurons degenerate, the muscles no longer receive signals from the brain and begin to waste away. ALS is characterized by stiff muscles, muscle twitching, and progressive muscle weakness from muscle wasting. The parts of the body affected by early symptoms of ALS depend on which motor neurons in the body are damaged first, usually the limbs. As the disease progresses most patients are unable to walk or use their arms and eventually develop difficulty speaking, swallowing and breathing. Most patients retain cognitive function and sensory neurons are generally unaffected. Patients are often diagnosed after the age of 40 and the median survival time from onset to death is around 3–4 years. In the final stages, patients can lose voluntary control of eye muscles and often die of respiratory failure or pneumonia as a result of degeneration of the motor neurons and muscles required for breathing. Currently there is no cure for ALS, only treatments that may prolong life.
Genetics and Underlying Causes
To date, multiple genes and proteins have been implicated in ALS. One of the common themes between many of these genes and their causative mutations is the presence of protein aggregates in motor neurons (Dewey et al., 2012). Other common molecular features in ALS patients are altered RNA metabolism (Polymenidou et al., 2012) and general histone hypoacetylation (Rouaux et al., 2003).

- The SOD1 gene on chromosome 21 that codes for the superoxide dismutase protein is associated with 2% of cases and is believed to be transmitted in an autosomal dominant manner (Battistini et al., 2009; Anderson et al., 1996). Many different mutations in SOD1 have been documented in ALS patients with varying degrees of progressiveness (Anderson et al., 1996). SOD1 protein is responsible for destroying naturally occurring, but harmful superoxide radicals produced by the mitochondria. Most of the SOD1 mutations associated with ALS are gain-of-function mutations in which the protein retains its enzymatic activity, but aggregate in motor neurons causing toxicity (Bruijn et al., 1998; Furukawa et al., 2006). Normal SOD protein is also implicated in other cases of ALS due to potentially cellular stress (Boillee et al., 2006). Researchers have developed an ALS mouse model through gain-of-function mutations in SOD1 (Cudkowicz et al., 1997).
- Recently a gene called c9orf72 was found to have a hexanucleotide repeat in the non-coding region of the gene in association with ALS and ALS-FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Majounie et al., 2012). These hexanucleotide repeats may be present in up 40% of familial ALS cases and 10% of sporadic cases. C9orf72 likely functions as a guanine exchange factor for a small GTPase, but this is likely not related to the underlying cause of ALS (Yoshimura et al., 2010). The hexanucleotide repeats are likely causing cellular toxicity after they are spliced out of the c9orf72 mRNA transcripts and accumulate in the nuclei of affected cells (Lee et al., 2013).
- The UBQLN2 gene encodes the protein ubiquilin 2 which is responsible for controlling the degradation of ubiquitinated proteins in the cell. Mutations in UBQLN2 interfere with protein degradation resulting in neurodegeneration through abnormal protein aggregation (Han-Xiang et al., 2011). This form of ALS is X chromosome-linked and dominantly inherited and can also be associated with dementia.
Epigenetic Treatment with HDAC inhibitors
ALS patients and mouse models show general histone hypoacetylation that can ultimately trigger apoptosis of cells (Rouaux et al., 2004). In experiments with mice, HDAC inhibtors counteract this hypoacetylation, reactivate aberrantly down-regulated genes, and counteract apoptosis initiation (Ryu et al., 2005; Yoo et al., 2011). Furthermore, HDAC inhibitors are known to prevent SOD1 protein aggregates in vitro (Corcoran et al., 2004).
- Sodium phenylbutyrate treatment in a SOD1 mouse model of ALS showed improved motor performance and coordination, decreased neural atrophy and neural loss, and increased weight gain (Del Signore et al., 2009; Petri et al., 2006). Release of pro-apoptotic factors was also abrogated as well as a general increase in histone acetylation (Ryu et al., 2005). A human trial using phenylbuturate in ALS patients showed some increase in histone acetylation, but the study did not report whether ALS symptoms improved with treatment (Cudkowicz et al., 2009).
- Valproic acid in mice studies restored histone acetylation levels, increased levels of pro-survival factors, and mice showed improved motor performance (Crochemore et al., 2009). However, while the drug delayed the onset of ALS, it did not increase lifespan or prevent denervation (Rouaux et al., 2007). Human trials of valproic acid in ALS patients did not improve survival or slow progression (Piepers et al., 2009).
- Trichostatin A trials in mouse ALS models restored histone acetylation in spinal neurons, decreased axon demyelination, and increased survival of mice (Yoo et al., 2011).
Spinal Muscular Atrophy (SMA)
- Main Wiki article: Spinal Muscular Atrophy

Spinal muscular atrophy (SMA) is an autosomal recessive motor neuron disease caused by mutations in the SMN1 gene (Brzustowicz et al., 1990). Symptoms vary greatly with each subset of SMA and the stage of the disease. General symptoms include overall muscle weakness and poor muscle tone including extremities and respiratory muscles leading to difficulty walking, breathing, and feeding. Depending on the type of SMA, the disease can present itself from infancy through adulthood. As SMN protein generally promotes the survival of motor neurons, mutations in SMN1 results in slow degeneration motor neurons leading to progressive system-wide muscle wasting. Specifically, over time, decreased levels of SMN protein results in gradual death of the alpha motor neurons in the anterior horn of the spinal cord and brain. Muscles depend on connections to motor neurons and the central nervous system to stimulate muscle maintenance and therefore degeneration of motor neurons and subsequent denervation of muscles lead to loss of muscle control and muscle atrophy. The muscles of the lower extremities are often affected first followed by upper extremities and sometime the muscles of respiration and mastication. In general, proximal muscle are always affected more than distal muscle.
Genetic Cause
Spinal muscular atrophy is linked to genetic mutations in the SMN1 (Survival of Motor Neuron 1) gene. The SMN protein is widely expressed in neurons and serves many functions within neurons including spliceosome construction, mRNA axon transport, neurite outgrowth during development, and neuromuscular junction formation. The causal function loss in SMA is currently unknown.
SMN1 is located in a telomeric region of human chromosome 5 and also contains SMN2 in a centromeric region. SMN1 and SMN2 are nearly identical except for a single nucleotide change in SMN2 resulting in an alternative splicing site where intron 6 meets exon 8. This single base pair change leads to only 10-20% of SMN2 transcripts resulting in fully functional SMN protein and 80-90% of transcripts leading to a truncated protein that is rapidly degraded. Most SMA patients have 2 or more copies of the SMN2 gene with more copies resulting a decrease in disease severity (Prior et al., 2009). Most SMA patients have either point mutations or a deletion in exon 7 often leading to a protein product similar to the truncated and degraded version of the SMN2 protein. In SMA patients this small amount of functional SMN2 protein product allows for the some neurons to survive.
Epigenetic Treatment through SMN2 gene activation
Although SMA is not caused by an epigenetic mechanism, therapeutic drugs that target epigenetic marks may provide SMA patients with some relief, halting or even reversing the progression of the disease. As SMA patients with higher copy numbers of the SMN2 gene have less severe symptoms, researchers predicted that epigenetic drugs that increased SMN2 mRNA expression would increase the amount of functional SMN protein in neurons leading to a reduction in SMA symptoms. Histone deacetylase (HDAC) inhibitors are the main compounds that have been tested to increase SMN2 mRNA expression. Inhibiting HDACs would allow for hyperacetylation of the SMN2 gene loci theoretically resulting in an increase in SMN2 expression (Kernochan et al., 2005). Many of these HDAC inhibitors (HDACi) are first tested in mouse models of SMA created through a variety of mutations in the mouse SMN1 gene. If the mice show improvement and the drug does not cause very many side effects or toxicity, the drug may be used in human clinical trials. Human trials with all of the below HDAC inhibitors are extremely variable and often impacted by the patient's exact SMA subtype.
- Quisinostat(JNJ-26481585)
- Quisinostat is effective at low doses resulting in some improved neuromuscular function in mouse model of SMA, but survival was not increased (Schreml et al., 2012). No human trials have been conducted.
- Sodium butyrate was the first HDAC inhibitor tested in SMA mouse models. It prolonged SMA mouse life span by 35% and showed increased levels of SMN protein in spinal cord tissue (Mercuri et al., 2007; Andreassi et al., 2004). However, sodium butyrate has not been used in human trials to date.
- Sodium phenybutyrate increases SMN2 full length mRNA transcripts in cell culture but drug application must be repeated in order to maintain results (Andreassi et al., 2004). Human trials show mixed results with one study showing increased SMA transcript levels in blood and improved motor function (Brahe et al. 2005), but a larger trial showing no effects on disease progression or motor function (Mercuri et al., 2007).
- Valproic acid added to cells from SMA patients increased SMN2 mRNA and protein levels and that the drug directly activates SMN2 promoter (Sumner et al., 2003; Brichta et al., 2003). In a SMA mouse model, valproic acid was added to the drinking water and restored motor neuron density and increased motor neuron number over a period of 8 months (Tsai et al., 2006). Human trials are extremely variable showing increased SMN2 levels and increased muscle strength in some trials and absolutely no effects in other trials (Weihl et al., 2006; Piepers et al., 2010; Swoboda et al., 2010; Darbar et al., 2011).
- M344
- M344 is a benzamide that shows promising results in fibroblast cell culture and increases level of splicing factors known to modulate SMN2 transcripts, but the drug was determined toxic and research has not progressed to in vivo testing (Riessland et al., 2006)
- Trichostatin A treatment shows promising results in mice . In one study, Trichostatin A combined with extra nourishment in early onset mouse SMA models resulted in improved motor function and survival and delays progressive denervation of muscles (Narver et al., 2008). A second study in a SMA mouse model showed increased SMN2 transcripts with daily injections (Avila et al., 2007). No human trials have been conducted.
- Vorinostat (SAHA)
- Vorinostat is a second generation inhibitor that is fairly non-toxic and found to be effective in cell culture at low concentrations (Hahnan et al., 2006) and increases histone acetylation at the SMN2 promoter (Kernochan et al., 2005). In a SMA mouse model, SAHA treatment resulted in weight gain, increased SMN2 transcripts levels in muscles and spinal cord, and motor neuron loss and denervation were halted (Riessland et al., 2010). No human trials have been conducted.
Neurodegenerative Diseases of the Central Nervous System
Alzheimer’s Disease (AD)
- Main Wiki article: Alzheimer’s Disease
Alzheimer’s disease (AD) is the most prevalent form of dementia among the elderly. The disease is characterized behaviorally by chronic and progressive decline in cognitive function, beginning with short term memory loss, and neurologically by buildup of misfolded tau protein and associated neurofibrillary tangles, and by amyloid-beta senile plaques amyloid-beta senile plaques. Several genetic factors have been identified as contributing to AD, including mutations to the amyloid precursor protein (APP) and presenilins 1 and 2 genes, and familial inheritance of apolipoprotein E allele epsilon 4. In addition to these common factors, there are a number of other genes that have shown altered expression in Alzheimer's disease, some of which are associated with epigenetic factors.
Epigenetic Factors of Alzheimer’s Disease
- ncRNA
- Research has shown that a ncRNA that is encoded antisense from an intron within the beta-amyloid cleaving enzyme gene, BACE1, is involved in AD (Faghihi et al., 2008). This ncRNA, BACE1-AS (for antisense), which overlaps exon 6 of BACE1, is involved in increasing the stability of the BACE1 mRNA transcript. As that gene's name suggests, BACE1 is an enzymatic protein that cleaves the Amyloid Precursor Protein into the insoluble amyloid beta form, which then aggregates into senile plaques. With increased stability of BACE1 mRNA resulting from BACE1-AS, more BACE1 mRNA is available for translation into BACE1 protein.
- miRNA factors have not consistently been shown to play a role in progression of AD. miRNAs are involved in post-transcriptional gene silencing via inhibiting translation or involvement in RNAi pathways. Some studies have shown upregulation of miRNA-146a, which differentially regulates neuroimmune-related Interleukin-1R associated kinases IRAK1 and IRAK2 expression, in human AD brain, while other studies have shown upregulation or downregulation of miRNA-9 in brain (Bennett et al., 2015).
- DNA methylation
- In Alzheimer’s disease cases, global DNA hypomethylation and gene-specific hypermethylation has been observed, though findings have varied between studies, especially in studies of human brains. Hypothetically, global hypomethylation should be associated with global increases in transcription, since CpG islands are most prevalent in gene promoters; gene-specific hypermethylation, however, would indicate that these hypermethylated genes are repressed by the methylation marks. Generally, repressive hypermethylation of genes related to learning and memory has been observed in conjunction with derepressive hypomethylation of neuroinflammatory genes and genes related to pathological expression of Alzheimer's disease.

- One study has shown reduced methylation in the long-term memory-associated temporal neocortex neurons in monozygotic twins with Alzheimer’s disease compared to the healthy twin (Mastroeni et al., 2009). Global hypomethylation of CpG dinucleotides has also been observed in hippocampus (Chouliaras et al., 2013) and in entorhinal cortex layer II (Mastroeni et al., 2010) of human AD patients, both of which are susceptible to AD pathology. These results, found by probing with immunoassays, have been challenged by studies that interrogate DNA sequence by bisulfite sequencing, a CpG transformation technique which is sensitive to CpG methylation status, in which global hypomethylation has been observed (Bakulski et al., 2012; Rao et al., 2012).
- At the individual gene level, Rao et al. have reported hypomethylation and thus derepression of COX-2, inhibition of which reduces inflammation and pain, and hypermethylation of BDNF, a neurotrophic factor important for long-term memory. Expression of CREB, an activity-dependent transcription factor involved in regulating BDNF among many other genes, has also been shown to be hypermethylated, and thus repressed, in AD brains, further reducing BDNF transcription (Rao et al., 2012). Furthermore, synaptophysin (SYP), the major synaptic vesicle protein-encoding gene, has been shown to be hypermethylated and thus repressed, and transcription factor NF-κB, which is involved in immune signaling, has been shown to be hypomethylated and thus derepressed (Rao et al., 2012). Taken together, these results have elucidated a role for dysregulation of genes involved in learning and memory and synaptic transmission, as well as with immune response.
- Hypomethylation has been observed in promoters of presenilin 1 (Wang et al., 2008), GSK3beta, which phosphorylates tau protein (Nicolia et al., 2010), and BACE1 (Byun et al., 2012), an enzyme that cleaves APP into the amyloid-beta form, which in turn aggregates into insoluble senile plaques. Repressive hypermethylation caused by amyloid-beta has been observed at the promoter of NEP, the gene for neprilysin, which is the major amyloid-beta clearing enzyme in the brain (Chen et al., 2009). This repression of NEP could result in a feed-forward buildup of senile plaques; combined with the observed increase in AD brains of BACE1-AS and corresponding increases in BACE1 protein and amyloid beta (Faghihi et al., 2008; see above), multiple levels of epigenetic regulation may be involved in controlling amyloid-beta formation, clearance or aggregation, and senile plaque deposition.
- There may be some effect of age in levels of DNA methylation at specific gene promoters, as one study found greater levels of methylation at APP promoters in AD patients up to 70 years old, but lower levels of methylation in patients greater than 70 years old (Tohgi et al., 1999). Studies on differential DNA methylation in human AD brains remain largely inconclusive possibly owing to the high degree of variability between individuals and to the numerous combinations of factors that may lead to AD.
- Histone marks
- Acetylation of lysine residues on histone tails is typically associated with transcriptional activation, whereas deacetylation is associated with transcriptional repression. There are few studies investigating specific histone marks in AD. These studies have elucidated a decrease in acetylation of lysines 18 and 23 on N-terminal tails of histone 3 (H3K18 and H3K23, respectively) (Zhang et al., 2012) and increases in HDAC2 in AD brains (Graff et al., 2012) - both marks related to transcriptional repression. Age-related cognitive decline has been associated with deregulation of H4K12 acetylation, a cognitive effect that was restored in mice by induction of this mark (Peleg et al., 2010).
Treatments for epigenetic factors of Alzheimer’s disease Treatment for prevention or management of Alzheimer's disease has proven troublesome since the disease is chronic and progressive, and many epigenetic drugs act globally and not in a gene-specific manner. As with other potential treatments to prevent or ameliorate symptoms of AD, these therapies do not work to cure, but only ameliorate symptoms of the disease temporarily, underscoring the chronic, progressive nature of AD, and the variability of methylation in AD brains.
DNA methylation-directed treatments
- Folate and other B vitamins
- B vitamins are involved in the metabolic pathway that leads to SAM production. SAM is the donor of the methyl group utilized by DNA methyltransferases (DNMTs) to methylate CpGs. Using animal models, Fuso et al. have demonstrated restoration of methylation at previously hypomethylated promoters of presenilin 1, BACE1 and APP (Fuso et al., 2005, 2008, 2012) - a hypothetically stable epigenetic modification that should repress those genes and slow the progression of AD. Dietary SAM supplementation has also been shown to reduce oxidative stress and delay buildup of neurological hallmarks of AD such as amyloid beta and phosphorylated tau protein in transgenic AD mice.
- AZA
- Khan and colleagues have demonstrated a potential role for neuroglobinin attenuating amyloid-related neurotoxicity (Khan et al., 2007). 5-aza-2' deoxycitidine (AZA, or decitabine), a DNMT inhibitor, has shown some evidence for regulating neuroglobin expression, though this finding has not been tested in AD models (Zhang et al., 2011).
Histone-directed treatments
- Though studies of histone marks in AD brains are few in number, several studies have looked at the effects of HDACis in treatment of Alzheimer’s disease. Class I and II HDAC inhibitors such as trichostatin A, vorinostat, and sodium butyrate, and Class III HDACis, such as nicotinamide, have been effective at treating symptoms in animal models of AD. While promising as a therapeutic in animal models, studies on the long-term efficacy of HDACis and human trials have yet to be conducted.
- Sodium butyrate
- Sodium butyrate is a class I and II HDACi and has been shown to recover learning and memory after 4 weeks (Fischer et al., 2007), decrease phosphorylated tau protein, and restore dendritic spine density in the hippocampus of AD transgenic mice (Ricobaraza et al., 2010). Histone acetylation resulting from diffuse sodium butyrate application is especially prevalent in the hippocampus, and genes involved in learning and memory showed increased acetylation in AD mice treated with this drug (Govindarajan et al., 2011).
- Trichostatin A
- Trichostatin A is also a class I and II HDACi that rescues fear learning in a fear conditioning paradigm in transgenic AD mice to wild type levels via acetylation on histone 4 lysine tails (Francis et al., 2009)
- Vorinostat
- Vorinostat is a class I and II HDACi that has been shown to be especially effective at inhibiting HDAC2 and restoring memory functions in non-AD models of learning deficits (Guan et al., 2009). One study showed vorinostat is effective at reversing contextual memory deficits in transgenic AD mice (Kilgore et al., 2010).
Huntington's (HD)
- Main Wiki article: Huntington's disease
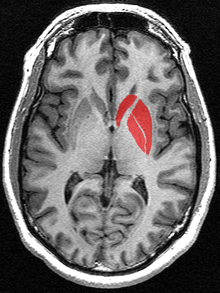
Huntington’s disease (HD) is an inherited disorder that causes progressive degeneration of neurons within the cerebral cortex and striatum of the brain (OMIM 143100) resulting in loss of motor functions (involuntary muscle contractions), decline in cognitive ability (eventually resulting in dementia), and changes in behavior (Urdinguio et al., 2009).
Genetics and Underlying Causes
Huntington’s is caused by an autosomal dominant mutation expanding the number of glutamine codon repeats (CAG) within the Huntingtin gene (Htt)(OMIN 613004) (http://www.omim.org/entry/143100). The Htt gene encodes for the huntingtin protein which plays a role in normal development but its exact function remains unknown (Nasir et al., 1995). The length of this CAG repeat correlates with the age-of-onset of the disease. The average person without Huntington’s has less than 36 CAG repeats present within the Htt gene. When this repeat length exceeds 36, the onset of neuronal degradation and the physical symptoms of Huntington’s can range from as early as 5 years of age (CAG repeat > 70) to as late as 80 years of age (CAG repeat < 39) (Chen et al., 2002).
This CAG expansion results in mRNA downregulation of specific genes, decreased histone acetylation, and increased histone methylation (Ryu et al., 2006; Hazeki et al., 2002). The exact mechanism of how this repeat causes gene dysregulation is unknown, but epigenome modification may play a role. For early-onset Huntington's (ages 5–15), both transgenic mice and mouse striatal cell lines show brain specific histone H3 hypoacetylation and decreased histone association at specific downregulated genes within the striatum (namely Bdnf, Cnr1, Drd2 - dopamine 2 receptor, and Penk1 - preproenkephalin) (Sadri et al., 2007). For both late- and early-onset Huntington’s, the H3 and H4 core histones associated with these downregulated genes in Htt mutants have hypoacetylation (decreased acetylation) compared to wild-type Htt (Hazeki et al., 2002; Sadri et al., 2007). This hypoacetylation is sufficient to cause tighter chromatin packing and mRNA downregulation (Hazeki et al., 2002).
Along with H3 hypoacetylation, both human patients and mice with the mutant Htt have increased levels of histone H3 lysine 9 trimethylation (Ryu et al., 2006). This increase in H3-K9 trimethylation is linked to an increased expression of the methyltransferase ESET/SETDB1 (ERG-associated protein with SET domain (ESET)), which targets and trimethylates H3-K9 residues (Ryu et al., 2006). It’s proposed that this hypermethylation may account for the onset of specific gene repression in Htt mutants (Ryu et al., 2006).
Epigenetic Treatment with HDAC inhibitors
Huntington patients and both mouse and Drosophila models show histone H3 and H4 hypoacetylation. There are currently no treatments for the disease but numerous HDAC inhibitors have been tested and shown to reverse the certain symptoms caused by the Htt mutation.
- Sodium Butyrate
- Sodium butyrate treatment slowed neuronal degeneration in Drosophila models (Steffan et al., 2001). Sodium butyrate treatment also increased histone H3 acetylation and normalized mRNA levels for mutant Htt downregulated genes (Sadri et al., 2007).
- Valproic acid
- Valproic acid treatment increased mutant Htt H3 and H4 acetylation levels comparable to wild-type Htt in Drosophila models (Steffan et al., 2001).
- Sodium Phenylbutyrate
- Sodium phenylbutyrate phase II human triasl with 12 to 15 g/day showed restored mRNA levels of Htt mutant repressed genes but also had adverse side effects such as nausea, headaches, and gain instability (Hogarth et al., 2007). Phenylbutyrate has also been shown to increase histone acetylation, decrease histone methylation, increase survival rate, and decrease the rate of neuronal degradation in Htt mutant mouse models (Gardian et al., 2005).
- Trichostatin A
- Trichostatin A (TSA) treatment increased mutant Htt H3 and H4 acetylation levels comparable to wild-type Htt in Drosophila models (Steffan et al., 2001). TSA treatment has also been shown to increase alpha-tubulin lysine 40 acetylation in mouse striatal cells and increase intracellular transport of BDNF, a brain-derived neurotrophic factor that function in nerve growth and maintenance within the brain (Dompierre et al., 2007; http://ghr.nlm.nih.gov/gene/BDNF).
- Vorinostat (SAHA)
- Vorinostat treatment slowed photoreceptor degeneration and improved longevity of adult Htt mutant Drosophila (Steffan et al., 2001). Like TSA, SAHA treatment increased alpha-tubulin lysine 40 acetylation in mouse striatal cells and also increased intracellular transport of BDNF.
Parkinson's Disease (PD)
- Main Wiki article: Parkinson's Disease
Parkinson’s disease (PD) is characterized by progressive degeneration of dopaminergic neurons in the substantia nigra by causes unknown. Several genes and environmental factors (e.g. pesticide exposure) may play a role in onset of PD. Hallmarks include mutations to the alpha-synuclein gene, SNCA, as well as PARK2, PINK1, UCHL1, DJ1, and LRRK2 genes, and fibrillar accumulation of Lewy bodies from misfolded alpha-synuclein. Symptoms are most noticeably manifested in disorders of movement, including shaking, rigidity, deficits in making controlled movements, and slow and difficult walking. The late stages of the disease result in dementia and depression. Levodopa and dopaminergic therapy may ameliorate symptoms, though there is no treatment to halt progression of the disease.
Epigenetic factors of Parkinson’s disease
- ncRNA
- Research has shown reductions of miR-133b correlated to decreased numbers of dopaminergic neurons in the midbrain of PD patients (Kim et al., 2007). miR-132, meanwhile, is negatively correlated with dopaminergic neuron differentiation in the midbrain (Jankovic et al., 2005). miR-7 and miR-153 act to reduce alpha-synuclein levels (a hallmark of PD) but are reduced in PD brain (Doxakis, 2010).
- DNA methylation
- Neurons of PD patients show hypomethylation of tumor necrosis factor (TNF) alpha encoding sequence, overexpression of which leads to apoptosis of neurons (Pieper et al., 2008). Cerebrospinal fluid of PD patients also shows elevated TNF alpha (Mogi et al., 1996). Research indicates there may be a link between DNA methylation and SNCA expression (Bonsch et al., 2005; Jowaed et al., 2010). Furthermore, human and mouse models have shown reduction of nuclear DNMT1 levels in PD subjects, resulting in hypomethylated states associated with transcriptional repression (Desplats et al., 2011).
- Histone marks
- alpha-synuclein, the protein encoded by SNCA, can associate with histones and prevent their acetylation in concert with the HDACs HDAC1 and Sirt2 (Kontopoulos et al., 2006; Outeiro et al., 2007). Furthermore, it has been demonstrated that alpha-synuclein binds histone 3 and inhibits its acetylation in Drosophila (Kontopoulos et al., 2006). Dopamine depletion in Parkinson’s disease is associated with repressive histone modifications, including reduced H3K4me3, and lower levels of H3 and H4 lysine acetylation after levodopa therapy (a common treatment of PD).
Treatments for epigenetic factors of Parkinson’s disease Epigenetic treatments tested in models of PD are few, though some promising research has been conducted. Most treatments investigated thus far are directed at histone modifications and analysis of their roles in mediating alpha-synuclein expression and activity. Pesticides and paraquat increase histone acetylation, producing neurotoxic effects similar to those seen in PD, such as apoptosis of dopaminergic cells (Song et al., 2010, 2011). Despite this, treatment with HDACis (Harrison and Dexter, 2013) seems to have a neuroprotective effect.
- Sodium butyrate
- Several studies using different animal models have demonstrated that sodium butyrate may be effective in reducing alpha-synuclein-related neurotoxicity(Zhou et al., 2011; Rane et al., 2012). In Drosophila, sodium butyrate improved locomotor impairment and reduced early mortality rates (St. Laurent et al., 2013).
- Valproic acid
- In an inducible rat model of PD, valproic acid had a neuroprotective effect by preventing translocation of alpha-synuclein into cell nuclei (Monti et al., 2010).
- Vorinostat
- In an alpha-synuclein overexpressing Drosophila model of PD, vorinostat (as well as sodium butyrate) reduced alpha-synuclein-mediated neurotoxicity (Kontopolous et al., 2006).
- siRNA inhibition of SIRT2
- Treatment with SIRT2 inhibiting siRNA leads to reduced alpha-synuclein neurotoxicity AK-1 or AGK-2 (Outeiro et al., 2007).
See also
Notes
References
- Sghirlanzoni, Angelo, Davide Pareyson, and Giuseppe Lauria. "Sensory neuron diseases." The Lancet Neurology 4.6 (2005): 349-361.
- Goll, Mary Grace, and Timothy H. Bestor. "Eukaryotic cytosine methyltransferases." Annu. Rev. Biochem. 74 (2005): 481-514.
- Bernstein, Bradley E., Alexander Meissner, and Eric S. Lander. "The mammalian epigenome." Cell 128.4 (2007): 669-681.
- Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G 3rd, Kenny PJ, Wahlestedt C. "Expression of a noncoding RNA is elevated in Alzheimer's disease and drives rapid feed-forward regulation of beta-secretase." Nat Med. 14.7 (2008): 723-30.
- Urdinguio, Rocio G, Jose V Sanchez-Mut, and Manel Esteller. “Epigenetic Mechanisms in Neurological Diseases: Genes, Syndromes, and Therapies.” The Lancet Neurology 8, no. 11 (November 2009): 1056–72. doi:10.1016/S1474-4422(09)70262-5.
- Peedicayil, Jacob. “Epigenetic Drugs for Alzheimer’s Disease.” British Journal of Clinical Pharmacology 75, no. 4 (April 2013): 1152–53. doi:10.1111/j.1365-2125.2012.04444.x.
- Boillee S, Vande Velde C, Cleveland DW. “ALS: a disease of motor neurons and their nonneuronal neighbors.” Neuron 52 (2006): 39-59.
- Battistini S, Ricci C, Lotti EM, Benigni M, Gagliardi S, Zucco R, Bondavalli M, Marcello N, Ceroni M, Cereda C. "Severe familial ALS with a novel exon 4 mutation (L106F) in the SOD1 gene". J Neurol Sci. 293.1 (2010): 112–115.
- Andersen PM, Forsgren L, Binzer M, Nilsson P, Ala-Hurula V, Keränen ML, Bergmark L, Saarinen A, Haltia T, Tarvainen I, Kinnunen E, Udd B, Marklund SL. "Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation, A clinical and genealogical study of 36 patients". Brain. 119.4 (1996): 1153–1172.
- Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, Hayden DL, Schoenfeld DA, Hosler BA, Horvitz HR, Brown RH. "Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis." Ann Neurol. 41.2 (1997): 210–21.
- Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attiq J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, Adachi Y, Sardone V, Miller JW, Smith BN, Gallo JM, Ule J, Hirth F, Rogelj B, Houart C, Shaw CE. “Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins and are neurotoxic.” Cell Rep. 5.5 (2013): 11178-86.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Peterson RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. “Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS.” Neuron. 72 (2011): 245-256.
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. “A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD.” Neuron. 72 (2011): 257-268.
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ. "Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study." Lancet Neurol. 11.4 (2012): 323–30.
- Yoshimura S, Gerondopoulos A, Linford A, Rigden DJ, Barr FA. "Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors". J Cell Biol. 191.2 (2010): 367–81.
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. "Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1." Science. 281.5384 (1998): 1851–4.
- Han-Xiang Deng, Wenjie Chen, Seong-Tshool Hong, et al. "Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia". Nature. 477.7363 (2011): 211–215.
- Furukawa Y, Fu R, Deng HX, Siddique T, O'Halloran TV. "Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice." Proc Natl Acad Sci USA. 103.18 (2006): 7148–53.
- Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G. “TDP-43 aggregation in neurodegeneration: are stress granules the key?” Brain Res. 1462 (2012): 16-25.
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Bennett CF, Cleveland DW, Yeo GW. “Misregulated RNA processing in amyotrophic lateral sclerosis.” Brain Res. 1462 (2012): 3-15.
- Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL. “Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration.” EMBO J. 22 (2003): 6537-49.
- Corcoran LJ, Mitchison TJ, Liu Q. “A novel action of histone deacetylase inhibitors in a protein aggresome disease model.” Curr Biol. 14 (2004): 488-92.
- Rouaux C, Loeffler JP, Boutillier AL. “Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders.” Biochem Pharmacol. 68 (2004): 1157-64.
- Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Gangond F, Cormier KA, Cudkowicz ME, Brown RH, Ferrante RJ. “Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice.” J Neurochem. 93 (2005):1087-98.
- Del Signore SJ, Amante DJ, Kim J, Stack EC, Goodrich S, Cormier K, Smith K, Cudkowicz ME, Ferrante RJ. “Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice.” Amyotroph Lateral Scler. 10 (2009): 85-94.
- Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. “Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis.” Neurobiol Dis. 22 (2006): 40-9.
- Cudkowicz ME, Andres PL, Macdonald SA, Bedlack RS, Choudry R, Brown RH, Zhang H, Schoenfeld DA, Shefner J, Matson S, Matson WR, Ferrante RJ. “Phase 2 study of sodium phenylbutyrate in ALS.” Amyotroph Lateral Scler. 10 (2009):99-106.
- Crochemore C, Virgili M, Bonamassa B, Canistro D, Pena-Altamira E, Paollini M, Contestabile A.” Long-term dietary administration of valproic acid does not affect, while retinoic acid decreases, the lifespan of G93A mice, a model for amyotrophic lateral sclerosis.” Muscle Nerve. 39 (2009): 548-52.
- Piepers S, Veldink JH, de Jong SW, van der Tweel I, van der Pol WL, Uijtendaal EV, Schelhaas HJ, Scheffer H, de Visser M, de Jong JM, Wokke JH, Groeneveld GJ, van den Berg LH. “Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis.” Ann Neurol. 66 (2009): 227-34.
- Yoo YE, Ko CP. “Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis.” Exp Neurol. 231 (2011): 147-59.
- Brichta L, Hofmann, Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B. "Valproic acid increases the SMN2 protein level: A well-known drug as a potential therapy for spinal muscular atrophy." Hum Mol Genet. 12.19 (2003): 2481–2489.
- Schreml J, Riessland M, Paterno M, Garbes L, Robbach K, Ackerman B, Kramer J, Somers E, Parson SH, Heller R, Berkessel A, Sterner-Kock A, Wirth B. “Severe SMA mice show organ impairment that cannot be rescued by therapy with the HDACi JNJ-26481585.” Eur J Hum Genet. 21.6 (2012): 643-52.
- Riessland M, Ackermann B, Forster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, Wirth B. “SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy.” Hum Mol Genet. 19.8 (2010): 1492-506.
- Narver HL, Kong L, Burnett BG, Choe DW, Bosch-Marce M, Taye AA, Eckhaus MA, Sumner CJ. “Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition.” Ann Neurol. 64 (2008): 465-70.
- Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgeman SJ, Burghes AH, Kissel JT. “A positive modifier of spinal muscular atrophy in the SMN2 gene.” Am J Hum Genet. 85.3 (2009): 408-413.
- Hahnen E, Eyupoglu IY, Brichta L, Haastert K, Trankle C, Siebzehnrubl FA, Riessland M, Holker I, Claus P, Romstock J, Buslei R, Wirth B, Blumcke I. “In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy.” J Neurochem. 98 (2006): 193-202.
- Brzustowicz LM, Lehner T, Castilla LH, Penchaszadeh GK, Wilhelmsen KC, Daniels R, Davies KE, Leppert M, Ziter F, Wood D, Dubowitz V, Zerres K, Hausmanowa-Petrusewicz I, Ott J, Munsat TL, Gilliam TC. "Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2–13.3." Nature 344.6266 (1990): 540–541.
- Su YN, Hung CC, Lin SY, Chen F, Chern JPS, Tsai C, Chang TS, Yang CC, Li H, Ho HN, Lee CN. "Carrier Screening for Spinal Muscular Atrophy (SMA) in 107,611 Pregnant Women during the Period 2005–2009: A Prospective Population-Based Cohort Study." PLoS ONE 6.2 (2011): e17067.
- Kernochan LE, Russo ML, Woodling NS, Huynh TN, Avila AM, Fischbeck KH, Sumner CJ. The role of histone acetylation in SMN gene expression. Hum Mol Genet. 14 (2005): 1171-82.
- Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH, Li H. “Treatment of spinal muscular atrophy by sodium butyrate.” Proc Natl Acad Sci USA. 98 (2001): 9808-13.
- Andreassi C, Angelozzi C, Tiziano FD, Vitali T, De Vincenzi E, Boninsegna A, Villanova M, Bertini E, Pini A, Neri G, Brahe C. "Phenylbutyrate increases SMN expression in vitro: Relevance for treatment of spinal muscular atrophy." Eur J of Hum Genet. 12.1 (2003): 59–65.
- Brahe C, Vitali T, Tiziano FD, Angelozzi C, Pinto AM, Borgo F, Moscato U, Bertini E, Mercuri E, Neri G. "Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients." Eur J of Hum Genet. 13.2 (2004): 256–259.
- Mercuri E, Bertini E, Messina S, Solari A, d'Amico A, Angelozzi C, Battini R, Berardinelli A, Boffi P, Bruno C, Cini C, Colitto F, Kinali M, Minetti C, Mongini T, Morandi, L, Neri G, Orcesi S, Pane M, Pelliccioni, Pini A, Tiziano FD, Villanova M, Vita G, Brahe C. "Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy." Neurology 68.1 (2007): 51–55.
- Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD< Schussler K, Chen X, Jarecki J, Burghes AH, Taylor JP, Fischbeck KH. “Valproic acid increases SMN levels in spinal muscular atrophy patient cells.” Ann Neurol. 54 (2003): 647-54.
- Riessland M, Brichta L, Hahnen E, Wirth B. "The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells." Hum Genet. 120.1 (2006): 101–110.
- Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. “Trichostatin A increases SMN expression and survival in a mouse model of SMA.” J Clin Invest. 117.3 (2007): 659-71.
- Weihl CC, Connolly AM, Pestronk A. “Valproate may improve strength and function in patients with type III/IV spinal muscle atrophy.” 8.67 (2006): 500-1.
- Piepers S, Cobben JM, Sodaar P, Jansen MD, Wadman RI, Meester-Delver A, Poll-The BT, Lemmick HH, Wokke JH, van der Pol WL, van den Berg LH.. “Quantification of SMN protein in leucocytes from spinal muscular atrophy patients: effects of treatment with valproic acid.” J Neurol Neurosurg Psychiatry. 82.8 (2010): 850-2.
- Swoboda KJ, Scott CB, Crawford TO, Simard LR, Reyna SP, Krosschell KJ, Acsadi G, Elsheik B, Schroth MK, D’Anjou G, LaSalle B, Prior TW, Sorenson SL, Maczulski JA, Bromberg MB, Chan GM, Kissel JT. “SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy.” PLoS One. 5 (2010): e12140.
- Darbar IA, Plaggert PG, Resende MB, Zanoteli E, Reed UC. “Evaluation of muscle strength and motor abilities in children with type II and III spinal muscle atrophy treated with valproic acid.” BMC Neurol. 11 (2011): 36.
- Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G 3rd, Kenny PJ, Wahlestedt C. "Expression of a noncoding RNA is elevated in Alzheimer's disease and drives rapid feed-forward regulation of beta-secretase." Nat Med. 14.7 (2008): 723-30.
- Bennett DA, Yu L, Yang J, Srivastava GP, Aubin C, De Jager PL. "Epigenomics of Alzheimer's disease." Transl Res. 165.1 (2015): 200-20.
- Mastroeni D, McKee A, Grover A, Rogers J, Coleman PD. "Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer's disease." PLoS One 4.8 (2009): e6617.
- Chouliaras L, Mastroeni D, Delvaux E, Grover A, Kenis G, Hof PR, Steinbusch HW, Coleman PD, Rutten BP, van den Hove DL. "Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients." Neurobiol Aging 34.9 (2013): 2091-9.
- Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. "Epigenetic changes in Alzheimer's disease: decrements in DNA methylation." Neurobiol Aging. 31.12 (2010): 2025-37.
- Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, Albin RL, Hu H, Rozek LS. "Genome-wide DNA methylation differences between late-onset Alzheimer's disease and cognitively normal controls in human frontal cortex." J Alzheimers Dis. 29.3 (2012): 571-88.
- Rao JS, Keleshian VL, Klein S, Rapoport SI. "Epigenetic modifications in frontal cortex from Alzheimer's disease and bipolar disorder patients." Transl Psychiatry. 2 (2012): e132.
- Wang Y, Zhang JX, Du XX, Zhao L, Tian Q, Zhu LQ, Wang SH, Wang JZ. "Temporal correlation of the memory deficit with Alzheimer-like lesions induced by activation of glycogen synthase kinase-3." J Neurochem. 106.6 (2008): 2364-74.
- Nicolia V, Fuso A, Cavallaro RA, Di Luzio A, Scarpa S. "B vitamin deficiency promotes tau phosphorylation through regulation of GSK3beta and PP2A." J Alzheimers Dis. 19.3 (2010): 895-907.
- Byun CJ, Seo J, Jo SA, Park YJ, Klug M, Rehli M, Park MH, Jo I. "DNA methylation of the 5'-untranslated region at +298 and +351 represses BACE1 expression in mouse BV-2 microglial cells." Biochem Biophys Res Commun. 417.1 (2012): 387-92.
- Chen KL, Wang SS, Yang YY, Yuan RY, Chen RM, Hu CJ. "The epigenetic effects of amyloid-beta(1-40) on global DNA and neprilysin genes in murine cerebral endothelial cells." Biochem Biophys Res Commun. 378.1 (2009): 57-61.
- Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C. "Alterations of 3-nitrotyrosine concentration in the cerebrospinal fluid during aging and in patients with Alzheimer's disease." Neurosci Lett. 269.1 (1999): 52-4.
- Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H, Kirsch W. "Targeted proteomics for quantification of histone acetylation in Alzheimer's disease." Proteomics 12.8 (2012): 1261-8.
- Gräff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai LH. "An epigenetic blockade of cognitive functions in the neurodegenerating brain." Nature 483.7388 (2012): 222-6.
- Peleg S, Sananbenesi F, Zovoilis A, Burkhardt S, Bahari-Javan S, Agis-Balboa RC, Cota P, Wittnam JL, Gogol-Doering A, Opitz L, Salinas-Riester G, Dettenhofer M, Kang H, Farinelli L, Chen W, Fischer A. "Altered histone acetylation is associated with age-dependent memory impairment in mice." Science 328.5979 (2010): 753-6.
- Fuso A, Seminara L, Cavallaro RA, D'Anselmi F, Scarpa S. "S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production." Mol Cell Neurosci. 28.1 (2005): 195-204.
- Fuso A, Nicolia V, Cavallaro RA, Ricceri L, D'Anselmi F, Coluccia P, Calamandrei G, Scarpa S. "B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice." Mol Cell Neurosci. 37.4 (2008): 731-46.
- Fuso A, Cavallaro RA, Nicolia V, Scarpa S. "PSEN1 promoter demethylation in hyperhomocysteinemic TgCRND8 mice is the culprit, not the consequence." Curr Alzheimer Res. 9.5 (2012): 527-35.
- Khan AA, Mao XO, Banwait S, Jin K, Greenberg DA. "Neuroglobin attenuates beta-amyloid neurotoxicity in vitro and transgenic Alzheimer phenotype in vivo." Proc Natl Acad Sci U S A. 104.48 (2007): 19114-9.
- Zhang W, Tian Z, Sha S, Cheng LY, Philipsen S, Tan-Un KC. "Functional and sequence analysis of human neuroglobin gene promoter region." Biochim Biophys Acta. 1809.4-6 (2011): 236-44.
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. "Recovery of learning and memory is associated with chromatin remodelling." Nature 447.7141 (2007): 178-82.
- Ricobaraza A, Cuadrado-Tejedor M, Marco S, Pérez-Otaño I, García-Osta A. "Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease." Hippocampus 22.5 (2010): 1040-50.
- Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A. "Sodium butyrate improves memory function in an Alzheimer's disease mouse model when administered at an advanced stage of disease progression." J Alzheimers Dis. 26.1 (2011): 187-97.
- Francis YI, Fà M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, Arancio O. "Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease." J Alzheimers Dis. 18.1 (2009): 131-9.
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJF, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. "HDAC2 negatively regulates memory formation and synaptic plasticity." Nature 459.7243 (2009): 55-60.
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. "Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease." Neuropsychopharmacology 35.4 (2010): 870-80.
- Nasir, Jamal, et al. "Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes." Cell 81.5 (1995): 811-823.
- Chen, Songming, Frank A. Ferrone, and Ronald Wetzel. "Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation." Proceedings of the National Academy of sciences 99.18 (2002): 11884-11889.
- Ryu, Hoon, et al. "ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease." Proceedings of the National Academy of Sciences 103.50 (2006): 19176-19181.
- Hazeki, Noriko, Tadashi Tsukamoto, Ikuru Yazawa, Minami Koyama, Shunji Hattori, Iori Someki, Takeshi Iwatsubo, Koichiro Nakamura, Jun Goto, and Ichiro Kanazawa. “Ultrastructure of Nuclear Aggregates Formed by Expressing an Expanded Polyglutamine.” Biochemical and Biophysical Research Communications 294, no. 2 (June 7, 2002): 429–40. doi:10.1016/S0006-291X(02)00498-9.
- Sadri-Vakili, Ghazaleh, Bérengère Bouzou, Caroline L. Benn, Mee-Ohk Kim, Prianka Chawla, Ryan P. Overland, Kelly E. Glajch, et al. “Histones Associated with Downregulated Genes Are Hypo-Acetylated in Huntington’s Disease Models.” Human Molecular Genetics 16, no. 11 (June 1, 2007): 1293–1306. doi:10.1093/hmg/ddm078.
- Steffan, Joan S., Laszlo Bodai, Judit Pallos, Marnix Poelman, Alexander McCampbell, Barbara L. Apostol, Alexsey Kazantsev, et al. “Histone Deacetylase Inhibitors Arrest Polyglutamine-Dependent Neurodegeneration in Drosophila.” Nature 413, no. 6857 (October 18, 2001): 739–43. doi:10.1038/35099568.
- Hogarth, Penelope, Luca Lovrecic, and Dimitri Krainc. “Sodium Phenylbutyrate in Huntington’s Disease: A Dose-Finding Study.” Movement Disorders 22, no. 13 (October 15, 2007): 1962–64. doi:10.1002/mds.21632.
- Gardian, Gabriella, et al. "Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease." Journal of Biological Chemistry 280.1 (2005): 556-563.
- Dompierre, Jim P., et al. "Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation." The Journal of neuroscience 27.13 (2007): 3571-3583.
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. "A MicroRNA feedback circuit in midbrain dopamine neurons." Science 317.5842 (2007): 1220-4.
- Jankovic J, Chen S, Le WD. "The role of Nurr1 in the development of dopaminergic neurons and Parkinson's disease." Prog Neurobiol. 77.1-2 (2005): 128-38.
- Doxakis E. "Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153." J Biol Chem. 285.17 (2010): 12726-34.
- Pieper HC, Evert BO, Kaut O, Riederer PF, Waha A, Wüllner U. "Different methylation of the TNF-alpha promoter in cortex and substantia nigra: Implications for selective neuronal vulnerability." Neurobiol Dis. 32.3 (2010): 521-7.
- Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. "Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson's disease." Neurosci Lett. 211.1 (1996): 13-6.
- Bönsch D, Lenz B, Kornhuber J, Bleich S. "DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism." Neuroreport. 16.2 (2005):167-70.
- Jowaed A, Schmitt I, Kaut O, Wüllner U. "Methylation regulates alpha-synuclein expression and is decreased in Parkinson's disease patients' brains." J Neurosci. 30.18 (2010): 6355-9.
- Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, Adame A, Rockenstein E, Masliah E. "Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases." J Biol Chem. 286.11 (2011): 9031-7.
- Kontopoulos E, Parvin JD, Feany MB. "Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity." Hum Mol Genet. 15.20 (2006): 3012-23.
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. "Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease." Science 317.5837 (2007): 516-9.
- Song C, Kanthasamy A, Anantharam V, Sun F, Kanthasamy AG. "Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: relevance to epigenetic mechanisms of neurodegeneration." Mol Pharmacol. 77.4 (2010): 621-32.
- Song C, Kanthasamy A, Jin H, Anantharam V, Kanthasamy AG. "Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration." Neurotoxicology 32.5 (2011): 586-95.
- Harrison IF, Dexter DT. "Epigenetic targeting of histone deacetylase: terapeutic potential in Parkinson's disease?" Pharmacol Ther. 140.1 (2013): 34-52.
- Zhou W, Bercury K, Cummiskey J, Luong N, Lebin J, Freed CR. "Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease." J Biol Chem. 286.17 (2011): 14941-51.
- Rane P, Shields J, Heffernan M, Guo Y, Akbarian S, King JA. "The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD." Neuropharmacology 62.7 (2012): 2409-12.
- St Laurent R, O'Brien LM, Ahmad ST. "Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson's disease." Neuroscience 246 (2013): 382-90.
- Monti B, Gatta V, Piretti F, Raffaelli SS, Virgili M, Contestabile A. "Valproic acid is neuroprotective in the rotenone rat model of Parkinson's disease: involvement of alpha-synuclein." Neurotox Res. 17.2 (2010): 130-41.