Senescence

Senescence (/sɪˈnɛsəns/) (from Latin: senescere, meaning "to grow old," from senex) or biological aging (also spelled biological ageing) is the gradual deterioration of function characteristic of most complex lifeforms, arguably found in all biological kingdoms, that on the level of the organism increases mortality after maturation. The word "senescence" can refer either to cellular senescence or to senescence of the whole organism. It is commonly believed that cellular senescence underlies organismal senescence. The science of biological aging is biogerontology.
Senescence is not the inevitable fate of all organisms and can be delayed. The discovery, in 1934, that calorie restriction can extend lifespan 50% in rats, and the existence of species having negligible senescence and potentially immortal species such as Hydra, have motivated research into delaying and preventing senescence and thus age-related diseases. Organisms of some taxonomic groups (taxa), including some animals, even experience chronological decrease in mortality, for all or part of their life cycle.[1] On the other extreme are accelerated aging diseases, rare in humans. There is also the extremely rare and poorly understood "Syndrome X," whereby a person remains physically and mentally an infant or child throughout one's life.[2][3]
Even if environmental factors do not cause aging, they may affect it; in such a way, for example, overexposure to ultraviolet radiation accelerates skin aging. Different parts of the body may age at different rates. Two organisms of the same species can also age at different rates, so that biological aging and chronological aging are quite distinct concepts.
Albeit indirectly, senescence is by far the leading cause of death (other than in the trivially accurate sense that cerebral hypoxia, i.e., lack of oxygen to the brain, is the immediate cause of all human death). Of the roughly 150,000 people who die each day across the globe, about two thirds — 100,000 per day — die of age-related causes; in industrialized nations, moreover, the proportion is much higher, reaching 90%.[4]
There are a number of hypotheses as to why senescence occurs; for example, some posit it is programmed by gene expression changes, others that it is the cumulative damage caused by biological processes. Whether senescence as a biological process itself can be slowed down, halted or even reversed, is a subject of current scientific speculation and research.[5]
Cellular senescence
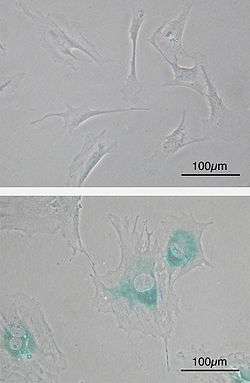
(upper) Primary mouse embryonic fibroblast cells (MEFs) before senescence. Spindle-shaped. (lower) MEFs became senescent after passages. Cells grow larger, flatten shape and expressed senescence-associated β-galactosidase (SABG, blue areas), a marker of cellular senescence.
Cellular senescence is the phenomenon by which normal diploid cells cease to divide. In culture, fibroblasts can reach a maximum of 50 cell divisions before becoming senescent. This phenomenon is known as "replicative senescence", or the Hayflick limit.[6] Replicative senescence is the result of telomere shortening that ultimately triggers a DNA damage response. Cells can also be induced to senesce via DNA damage in response to elevated reactive oxygen species (ROS), activation of oncogenes and cell-cell fusion, independent of telomere length. As such, cellular senescence represents a change in "cell state" rather than a cell becoming "aged" as the name confusingly suggests.
Although senescent cells can no longer replicate, they remain metabolically active and commonly adopt an immunogenic phenotype consisting of a pro-inflammatory secretome, the up-regulation of immune ligands, a pro-survival response, promiscuous gene expression (pGE) and stain positive for senescence-associated β-galactosidase activity.[7] The nucleus of senescent cells is characterized by senescence-associated heterochromatin foci (SAHF) and DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS).[8] Senescent cells affect tumour suppression, wound healing and possibly embryonic/placental development and a pathological role in age-related diseases.[9]
The experimental elimination of senescent cells from transgenic progeroid mice[10] and non-progeroid, naturally-aged mice[11][12][13] led to greater resistance against aging-associated diseases.
Epigenetic clock analysis of cellular senescence
According to a molecular biomarker of aging known as epigenetic clock,[14] the three major types of cellular senescence, namely replicative senescence, oncogene-induced senescence and DNA damage-induced senescence are distinct because induction of replicative senescence (RS) and oncogene-induced senescence (OIS) were found to be accompanied by epigenetic aging of primary cells but senescence induced by DNA damage was not, even though RS and OIS activate the cellular DNA damage response pathway.[15] These results highlight the independence of cellular senescence from epigenetic aging. Consistent with this, telomerase-immortalised cells continued to age (according to the epigenetic clock) without having been treated with any senescence inducers or DNA-damaging agents, re-affirming the independence of the process of epigenetic ageing from telomeres, cellular senescence, and the DNA damage response pathway. Although the uncoupling of senescence from cellular aging appears at first sight to be inconsistent with the fact that senescent cells contribute to the physical manifestation of organism ageing, as demonstrated by Baker et al., where removal of senescent cells slowed down aging .[10] However, the epigenetic clock analysis of senescence suggests that cellular senescence is a state that cells are forced into as a result of external pressures such as DNA damage, ectopic oncogene expression and exhaustive proliferation of cells to replenish those eliminated by external/environmental factors.[15] These senescent cells, in sufficient numbers, will undoubtedly cause the deterioration of tissues, which is interpreted as organism ageing. However, at the cellular level, aging, as measured by the epigenetic clock, is distinct from senescence. It is an intrinsic mechanism that exists from the birth of the cell and continues. This implies that if cells are not shunted into senescence by the external pressures described above, they would still continue to age. This is consistent with the fact that mice with naturally long telomeres still age and eventually die even though their telomere lengths are far longer than the critical limit, and they age prematurely when their telomeres are forcibly shortened, due to replicative senescence. Hence senescence is a route by which cells exit prematurely from the natural course of cellular ageing.[15]
Aging of the whole organism
Organismal senescence is the aging of whole organisms. In general, aging is characterized by the declining ability to respond to stress, increased homeostatic imbalance, and increased risk of aging-associated diseases. Death is the ultimate consequence of aging, though "old age" is not a scientifically recognized cause of death because there is always a specific proximal cause, such as cancer, heart disease, or liver failure. Aging of whole organisms is therefore a complex process that can be defined as "a progressive deterioration of physiological function, an intrinsic age-related process of loss of viability and increase in vulnerability."[16]
Differences in maximum life span among species correspond to different "rates of aging." For example, inherited differences in the rate of aging make a mouse elderly at 3 years and a human elderly at 80 years.[17] These genetic differences affect a variety of physiological processes, including the efficiency of DNA repair, antioxidant enzymes, and rates of free radical production.

Senescence of the organism gives rise to the Gompertz–Makeham law of mortality, which says that mortality rate accelerates rapidly with age.
Some animals, such as some reptiles and fish, age slowly (negligible senescence) and exhibit very long lifespans. Some even exhibit "negative senescence", in which mortality falls with age, in disagreement with the Gompertz–Makeham "law".[1]
Whether replicative senescence (Hayflick limit) plays a causative role in organismal aging is at present an active area of investigation.
The oft-quoted evolutionary theorist George Williams wrote, "It is remarkable that after a seemingly miraculous feat of morphogenesis, a complex metazoan should be unable to perform the much simpler task of merely maintaining what is already formed."[18]
There is a current debate as to whether or not the pursuit of longevity and the postponement of senescence are cost-effective health care goals given finite health care resources. Because of the accumulated infirmities of old age, bioethicist Ezekiel Emanuel, opines that the pursuit of longevity via the compression of morbidity hypothesis is a "fantasy" and that human life is not worth living after age 75; longevity then should not be a goal of health care policy.[19] This opinion has been refuted by neurosurgeon and medical ethicist Miguel Faria, who states that life can be worthwhile during old age, and that longevity should be pursued in association with the attainment of quality of life.[20] Faria claims that postponement of senescence as well as happiness and wisdom can be attained in old age in a large proportion of those who lead healthy lifestyles and remain intellectually active.[21]
Theories of aging
The exact etiology of senescence is still largely unclear and yet to be discovered. The process of senescence is complex, and may derive from a variety of different mechanisms and exist for a variety of different reasons. However, senescence is not universal. In a few simple species, such as those in the genus Hydra, senescence is negligible and cannot be detected.
All such species have no "post-mitotic" cells; they reduce the effect of damaging free radicals by cell division and dilution. Another related mechanism is that of the biologically immortal planarian flatworms, which have "apparently limitless [telomere] regenerative capacity fueled by a population of highly proliferative adult stem cells."[22] These organisms are biologically immortal but not immortal in the traditional sense as they are nonetheless susceptible to trauma and infectious and non-infectious disease. Moreover, average lifespans can vary greatly within and between species. This suggests that both genetic and environmental factors contribute to aging.
In general, theories that explain senescence have been divided between the programmed and stochastic theories of aging. Programmed theories imply that aging is regulated by biological clocks operating throughout the lifespan. This regulation would depend on changes in gene expression that affect the systems responsible for maintenance, repair, and defense responses. The reproductive-cell cycle theory suggests that aging is caused by changes in hormonal signaling over the lifespan.[23] Stochastic theories blame environmental impacts on living organisms that induce cumulative damage at various levels as the cause of aging, examples of which ranging from damage to DNA, damage to tissues and cells by oxygen radicals (widely known as free radicals countered by the even more well-known antioxidants), and cross-linking.
However, aging is seen as a progressive failure of homeodynamics–systemic preservation of homeostasis, involving genes for maintenance and repair, stochastic events leading to molecular damage and molecular heterogeneity, and chance events determining the probability of death. Since complex and interacting systems of maintenance and repair comprise the homeodynamic space of a biological system, aging is considered to be a progressive shrinkage of homeodynamic space mainly due to increased molecular heterogeneity. In 2013, a group of scientists defined nine hallmarks of aging that are common between organisms with emphasis on mammals: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication.[24]
Evolutionary theories
A gene can be expressed at various stages of life. Therefore, natural selection can support lethal and harmful alleles, if their expression occurs after reproduction. Senescence may be the product of such selection.[25][26][27] In addition, aging is believed to have evolved because of the increasingly smaller probability of an organism still being alive at older age, due to predation and accidents, both of which may be random and age-invariant. The antagonistic plietropy theory states that strategies which result in a higher reproductive rate at a young age, but shorter overall lifespan, result in a higher lifetime reproductive success and are therefore favoured by natural selection. In essence, aging is, therefore, the result of investing resources in reproduction, rather than maintenance of the body (the "Disposable Soma" theory[28]), in light of the fact that accidents, predation, and disease kill organisms regardless of how much energy is devoted to repair of the body. Various other theories of aging exist, and are not necessarily mutually exclusive.
The geneticist J. B. S. Haldane wondered why the dominant mutation that causes Huntington's disease remained in the population, and why natural selection had not eliminated it. The onset of this neurological disease is (on average) at age 45 and is invariably fatal within 10–20 years. Haldane assumed that, in human prehistory, few survived until age 45. Since few were alive at older ages and their contribution to the next generation was therefore small relative to the large cohorts of younger age groups, the force of selection against such late-acting deleterious mutations was correspondingly small. However, if a mutation affected younger individuals, selection against it would be strong. Therefore, late-acting deleterious mutations could accumulate in populations over evolutionary time through genetic drift, which has been demonstrated experimentally. This concept of higher accumulation of deleterious mutations for older organisms came to be known as the selection shadow.[29]
Peter Medawar formalised this observation in his mutation accumulation theory of aging.[30][31] "The force of natural selection weakens with increasing age—even in a theoretically immortal population, provided only that it is exposed to real hazards of mortality. If a genetic disaster... happens late enough in individual life, its consequences may be completely unimportant". The 'real hazards of mortality' are, in typical circumstances, predation, disease, and accidents. So, even an immortal population, whose fertility does not decline with time, will have fewer individuals alive in older age groups. This is called 'extrinsic mortality'. Young cohorts, not depleted in numbers yet by extrinsic mortality, contribute far more to the next generation than the few remaining older cohorts, so the force of selection against late-acting deleterious mutations, which affect only these few older individuals, is very weak. The mutations may not be selected against, therefore, and may spread over evolutionary time into the population.
The major testable prediction made by this model is that species that have high extrinsic mortality in nature will age more quickly and have shorter intrinsic lifespans. This is borne out among mammals, the best-studied in terms of life history. There is a correlation among mammals between body size and lifespan, such that larger species live longer than smaller species under controlled/optimum conditions, but there are notable exceptions. For instance, many bats and rodents are of similar size, yet bats live much longer. For instance, the little brown bat, half the size of a mouse, can live 30 years in the wild. A mouse will only live 2–3 years even under optimum conditions. The explanation is that bats have fewer predators, and therefore low extrinsic mortality. More individuals survive to later ages, so the force of selection against late-acting deleterious mutations is stronger. Fewer late-acting deleterious mutations equates to slower aging and therefore a longer lifespan. Birds are also warm-blooded and are similar in size to many small mammals, yet often live 5–10 times as long. They have less predation pressure than ground-dwelling mammals. Seabirds, which, in general, have the fewest predators of all birds, live longest.
When examining the body-size vs. lifespan relationship, one also observes that predatory mammals tend to live longer than prey mammals in a controlled environment, such as a zoo or nature reserve. The explanation for the long lifespans of primates (such as humans, monkeys, and apes) relative to body size is that their intelligence, and often their sociality, help them avoid becoming prey. High position in the food chain, intelligence and cooperativeness all reduce extrinsic mortality in species.
Another evolutionary theory of aging was proposed by George C. Williams[32] and involves antagonistic pleiotropy. A single gene may affect multiple traits. Some traits that increase fitness early in life may also have negative effects later in life. But, because many more individuals are alive at young ages than at old ages, even small positive effects early can be strongly selected for, and large negative effects later may be very weakly selected against. Williams suggested the following example: Perhaps a gene codes for calcium deposition in bones, which promotes juvenile survival and will therefore be favored by natural selection; however, this same gene promotes calcium deposition in the arteries, causing negative atherosclerotic effects in old age. Thus, harmful biological changes in old age may result from selection for pleiotropic genes that are beneficial early in life but harmful later on. In this case, selection pressure is relatively high when Fisher's reproductive value is high and relatively low when Fisher's reproductive value is low.
Gene regulation
A number of genetic components of aging have been identified using model organisms, ranging from the simple budding yeast Saccharomyces cerevisiae to worms such as Caenorhabditis elegans and fruit flies (Drosophila melanogaster). Study of these organisms has revealed the presence of at least two conserved aging pathways.
One of these pathways involves the gene Sir2, a NAD+-dependent histone deacetylase. In yeast, Sir2 is required for genomic silencing at three loci: the yeast mating loci, the telomeres and the ribosomal DNA (rDNA). In some species of yeast, replicative aging may be partially caused by homologous recombination between rDNA repeats; excision of rDNA repeats results in the formation of extrachromosomal rDNA circles (ERCs). These ERCs replicate and preferentially segregate to the mother cell during cell division, and are believed to result in cellular senescence by titrating away (competing for) essential nuclear factors. ERCs have not been observed in other species (nor even all strains of the same yeast species) of yeast (which also display replicative senescence), and ERCs are not believed to contribute to aging in higher organisms such as humans (they have not been shown to accumulate in mammals in a similar manner to yeast). Extrachromosomal circular DNA (eccDNA) has been found in worms, flies, and humans. The origin and role of eccDNA in aging, if any, is unknown.
Despite the lack of a connection between circular DNA and aging in higher organisms, extra copies of Sir2 are capable of extending the lifespan of both worms and flies (though, in flies, this finding has not been replicated by other investigators, and the activator of Sir2 resveratrol does not reproducibly increase lifespan in either species.[33]) Whether the Sir2 homologues in higher organisms have any role in lifespan is unclear, but the human SIRT1 protein has been demonstrated to deacetylate p53, Ku70, and the forkhead family of transcription factors. SIRT1 can also regulate acetylates such as CBP/p300, and has been shown to deacetylate specific histone residues.
RAS1 and RAS2 also affect aging in yeast and have a human homologue. RAS2 overexpression has been shown to extend lifespan in yeast.
Other genes regulate aging in yeast by increasing the resistance to oxidative stress. Superoxide dismutase, a protein that protects against the effects of mitochondrial free radicals, can extend yeast lifespan in stationary phase when overexpressed.
In higher organisms, aging is likely to be regulated in part through the insulin/IGF-1 pathway. Mutations that affect insulin-like signaling in worms, flies, and the growth hormone/IGF1 axis in mice are associated with extended lifespan. In yeast, Sir2 activity is regulated by the nicotinamidase PNC1. PNC1 is transcriptionally upregulated under stressful conditions such as caloric restriction, heat shock, and osmotic shock. By converting nicotinamide to niacin, nicotinamide is removed, inhibiting the activity of Sir2. A nicotinamidase found in humans, known as PBEF, may serve a similar function, and a secreted form of PBEF known as visfatin may help to regulate serum insulin levels. It is not known, however, whether these mechanisms also exist in humans, since there are obvious differences in biology between humans and model organisms.
Sir2 activity has been shown to increase under calorie restriction. Due to the lack of available glucose in the cells, more NAD+ is available and can activate Sir2. Resveratrol, a stilbenoid found in the skin of red grapes, was reported to extend the lifespan of yeast, worms, and flies (the lifespan extension in flies and worms have proved to be irreproducible by independent investigators[33]). It has been shown to activate Sir2 and therefore mimics the effects of calorie restriction, if one accepts that caloric restriction is indeed dependent on Sir2.
Gene expression is imperfectly controlled, and it is possible that random fluctuations in the expression levels of many genes contribute to the aging process as suggested by a study of such genes in yeast.[34] Individual cells, which are genetically identical, none-the-less can have substantially different responses to outside stimuli, and markedly different lifespans, indicating the epigenetic factors play an important role in gene expression and aging as well as genetic factors.
According to the GenAge database of aging-related genes there are over 700 genes associated with aging in model organisms: 555 in the soil roundworm (Caenorhabditis elegans), 87 in the bakers' yeast (Saccharomyces cerevisiae), 75 in the fruit fly (Drosophila melanogaster) and 68 in the mouse (Mus musculus).[35] The following is a list of genes connected to longevity through research on model organisms:
| Podospora | Saccharomyces | Caenorhabditis | Drosophila | Mus |
|---|---|---|---|---|
| grisea | LAG1 | daf-2 | sod1 | Prop-1 |
| LAC1 | age-1/daf-23 | cat1 | p66shc (Not independently verified) | |
| pit-1 | Ghr | |||
| RAS1 | daf-18 | mth | mclk1 | |
| RAS2 | akt-1/akt-2 | |||
| PHB1 | daf-16 | |||
| PHB2 | daf-12 | |||
| CDC7 | ctl-1 | |||
| BUD1 | old-1 | |||
| RTG2 | spe-26 | |||
| RPD3 | clk-1 | |||
| HDA1 | mev-1 | |||
| SIR2 | ||||
| aak-2 | ||||
| SIR4-42 | ||||
| UTH4 | ||||
| YGL023 | ||||
| SGS1 | ||||
| RAD52 | ||||
| FOB1 |
Cellular senescence
As noted above, senescence is not universal. It was once thought that senescence did not occur in single-celled organisms that reproduce through the process of cellular mitosis.[36] Recent investigation has unveiled a more complex picture. Single cells do accumulate age-related damage. On mitosis the debris is not evenly divided between the new cells. Instead it passes to one of the cells leaving the other cell pristine. With successive generations the cell population becomes a mosaic of cells with half ageless and the rest with varying degrees of senescence.[37]
Moreover, cellular senescence is not observed in several organisms, including perennial plants, sponges, corals, and lobsters. In those species where cellular senescence is observed, cells eventually become post-mitotic when they can no longer replicate themselves through the process of cellular mitosis; i.e., cells experience replicative senescence. How and why some cells become post-mitotic in some species has been the subject of much research and speculation, but (as noted above) it is sometimes suggested that cellular senescence evolved as a way to prevent the onset and spread of cancer. Somatic cells that have divided many times will have accumulated DNA mutations and would therefore be in danger of becoming cancerous if cell division continued. As such, it is becoming apparent that senescent cells undergo conversion to an immunogenic phenotype that enables them to be eliminated by the immune system.[38]
Lately, the role of telomeres in cellular senescence has aroused general interest, especially with a view to the possible genetically adverse effects of cloning. The successive shortening of the chromosomal telomeres with each cell cycle is also believed to limit the number of divisions of the cell, thus contributing to aging. There have, on the other hand, also been reports that cloning could alter the shortening of telomeres. Some cells do not age and are, therefore, described as being "biologically immortal". It is theorized by some that when it is discovered exactly what allows these cells, whether it be the result of telomere lengthening or not, to divide without limit that it will be possible to genetically alter other cells to have the same capability. It is further theorized that it will eventually be possible to genetically engineer all cells in the human body to have this capability by employing gene therapy and, therefore, stop or reverse aging, effectively making the entire organism potentially immortal.
The length of the telomere strand has senescent effects; telomere shortening activates extensive alterations in alternative RNA splicing that produce senescent toxins such as progerin, which degrades the tissue and makes it more prone to failure.[39]
Cancer cells are usually immortal. In about 85% of tumors, this evasion of cellular senescence is the result of up-activation of their telomerase genes.[40] This simple observation suggests that reactivation of telomerase in healthy individuals could greatly increase their cancer risk.
Ned Sharpless and collaborators demonstrated the first in vivo link between p16-expressing senescent cells and lifespan.[41] They found delayed senescent cell accumulation in mice with mutations that extend lifespan, as well as in mice that had their lifespan extended by food restriction. Later, Jan van Deursen and Darren Baker in collaboration with Andre Terzic at the Mayo Clinic in Rochester, Minn., provided the first in vivo evidence for a causal link between cellular senescence and aging by preventing the accumulation of senescent cells in BubR1 progeroid mice.[42] In the absence of senescent cells, the mice’s tissues showed a major improvement in the usual burden of age-related disorders. They did not develop cataracts, avoided the usual wasting of muscle with age. They retained the fat layers in the skin that usually thin out with age and, in people, cause wrinkling. Jan van Deursen, James Kirkland, Tamara Tchkonia, Nathan LeBrasseur, and Darren Baker at the Mayo Clinic in Rochester, Minn., provided the first direct in vivo evidence that cellular senescence causes signs of aging by eliminating senescent cells from progeroid mice by introducing a drug-inducible suicide gene and then treating the mice with the drug to kill senescent cells selectively, as opposed to decreasing whole body p16.[10] Another Mayo study led by James Kirkland in collaboration with Scripps and other groups demonstrated that senolytics, drugs that target senescent cells, enhance cardiac function and improve vascular reactivity in old mice, alleviate gait disturbance caused by radiation in mice, and delay frailty, neurological dysfunction, and osteoporosis in progeroid mice. Discovery of senolytic drugs was based on a hypothesis-driven approach: the investigators leveraged the observation that senescent cells are resistant to apoptosis to discover that pro-survival pathways are up-regulated in these cells. They demonstrated that these survival pathways are the "Achilles heel" of senescent cells using RNA interference approaches, including Bcl-2-, AKT-, p21-, and tyrosine kinase-related pathways. They then used drugs known to target the identified pathways and showed these drugs kill senescent cells by apoptosis in culture and decrease senescent cell burden in multiple tissues in vivo. Importantly, these drugs had long term effects after a single dose, consistent with removal of senescent cells, rather than a temporary effect requiring continued presence of the drugs. This was the first study to show that clearing senescent cells enhances function in chronologically aged mice.[43]
Chemical damage

One of the earliest aging theories was the Rate of Living Hypothesis described by Raymond Pearl in 1928[44] (based on earlier work by Max Rubner), which states that fast basal metabolic rate corresponds to short maximum life span.
While there may be some validity to the idea that for various types of specific damage detailed below that are by-products of metabolism, all other things being equal, a fast metabolism may reduce lifespan, in general this theory does not adequately explain the differences in lifespan either within, or between, species. Calorically restricted animals process as much, or more, calories per gram of body mass, as their ad libitum fed counterparts, yet exhibit substantially longer lifespans. Similarly, metabolic rate is a poor predictor of lifespan for birds, bats and other species that, it is presumed, have reduced mortality from predation, and therefore have evolved long lifespans even in the presence of very high metabolic rates.[45] In a 2007 analysis it was shown that, when modern statistical methods for correcting for the effects of body size and phylogeny are employed, metabolic rate does not correlate with longevity in mammals or birds.[46] (For a critique of the Rate of Living Hypothesis see Living fast, dying when?[47])
With respect to specific types of chemical damage caused by metabolism, it is suggested that damage to long-lived biopolymers, such as structural proteins or DNA, caused by ubiquitous chemical agents in the body such as oxygen and sugars, are in part responsible for aging. The damage can include breakage of biopolymer chains, cross-linking of biopolymers, or chemical attachment of unnatural substituents (haptens) to biopolymers.
Under normal aerobic conditions, approximately 4% of the oxygen metabolized by mitochondria is converted to superoxide ion, which can subsequently be converted to hydrogen peroxide, hydroxyl radical and eventually other reactive species including other peroxides and singlet oxygen, which can, in turn, generate free radicals capable of damaging structural proteins and DNA. Certain metal ions found in the body, such as copper and iron, may participate in the process. (In Wilson's disease, a hereditary defect that causes the body to retain copper, some of the symptoms resemble accelerated senescence.) These processes termed oxidative stress are linked to the potential benefits of dietary polyphenol antioxidants, for example in coffee,[48] red wine and tea.[49]
Sugars such as glucose and fructose can react with certain amino acids such as lysine and arginine and certain DNA bases such as guanine to produce sugar adducts, in a process called glycation. These adducts can further rearrange to form reactive species, which can then cross-link the structural proteins or DNA to similar biopolymers or other biomolecules such as non-structural proteins. People with diabetes, who have elevated blood sugar, develop senescence-associated disorders much earlier than the general population, but can delay such disorders by rigorous control of their blood sugar levels. There is evidence that sugar damage is linked to oxidant damage in a process termed glycoxidation.
Free radicals can damage proteins, lipids or DNA. Glycation mainly damages proteins. Damaged proteins and lipids accumulate in lysosomes as lipofuscin. Chemical damage to structural proteins can lead to loss of function; for example, damage to collagen of blood vessel walls can lead to vessel-wall stiffness and, thus, hypertension, and vessel wall thickening and reactive tissue formation (atherosclerosis); similar processes in the kidney can lead to renal failure. Damage to enzymes reduces cellular functionality. Lipid peroxidation of the inner mitochondrial membrane reduces the electric potential and the ability to generate energy. It is probably no accident that nearly all of the so-called "accelerated aging diseases" are due to defective DNA repair enzymes.
It is believed that the impact of alcohol on aging can be partly explained by alcohol's activation of the HPA axis, which stimulates glucocorticoid secretion, long-term exposure to which produces symptoms of aging.[50]
DNA damage theory
Alexander[51] was the first to propose that DNA damage is the primary cause of aging. Early experimental evidence supporting this idea was reviewed by Gensler and Bernstein.[52] By the early 1990s experimental support for this proposal was substantial, and further indicated that DNA damage due to reactive oxygen species was a major source of the DNA damages causing aging.[53][54][55][56][57] The current state of evidence bearing on this theory is reviewed in DNA damage theory of aging and by Bernstein et al.[58]
Reliability theory
Reliability theory suggests that biological systems start their adult life with a high load of initial damage. Reliability theory is a general theory about systems failure. It allows researchers to predict the age-related failure kinetics for a system of given architecture (reliability structure) and given reliability of its components. Reliability theory predicts that even those systems which are composed entirely of non-aging elements (with a constant failure rate) will nevertheless deteriorate (fail more often) with age, if these systems are redundant in irreplaceable elements. Aging, therefore, is a direct consequence of systems.
Reliability theory also predicts the late-life mortality deceleration with subsequent leveling-off, as well as the late-life mortality plateaus, as an inevitable consequence of redundancy exhaustion at extreme old ages. The theory explains why mortality rates increase exponentially with age (the Gompertz law) in many species, by taking into account the initial flaws (defects) in newly formed systems. It also explains why organisms "prefer" to die according to the Gompertz law, while technical devices usually fail according to the Weibull (power) law. Reliability theory allows to specify conditions when organisms die according to the Weibull distribution: Organisms should be relatively free of initial flaws and defects. The theory makes it possible to find a general failure law applicable to all adult and extreme old ages, where the Gompertz and the Weibull laws are just special cases of this more general failure law. The theory explains why relative differences in mortality rates of compared populations (within a given species) vanish with age (compensation law of mortality), and mortality convergence is observed due to the exhaustion of initial differences in redundancy levels.
Miscellanea
Biological clocks, which objectively measure the biological age of cells and tissues, may become useful for testing different biological aging theories.[14]
A set of rare hereditary (genetic) disorders, each called progeria, has been known for some time. Sufferers exhibit symptoms resembling accelerated aging, including wrinkled skin. The cause of Hutchinson–Gilford progeria syndrome was reported in the journal Nature in May 2003.[59] This report suggests that DNA damage, not oxidative stress, is the cause of this form of accelerated aging.
Recently, a kind of early senescence has been alleged to be a possible unintended outcome of early cloning experiments. The issue was raised in the case of Dolly the sheep, following her death from a contagious lung disease. The claim that Dolly's early death involved premature senescence has been vigorously contested,[60] and Dolly's creator, Dr. Ian Wilmut has expressed the view that her illness and death were probably unrelated to the fact that she was a clone.
See also
- Accelerated aging disease
- Advanced adult
- Ageing
- Aging brain
- Aging-associated disease
- Anti-aging medicine
- Anti-aging movement
- Biogerontology
- DNA damage theory of aging
- DNA repair
- Free radicals
- Genetics of aging
- Homeostatic capacity
- Immortality
- Indefinite lifespan
- List of life extension-related topics
- Mitohormesis
- Oxidative stress
- Phenoptosis
- Plant senescence
- Programmed cell death
- Regenerative medicine
- Rejuvenation (aging)
- SAGE KE
- Strategies for Engineered Negligible Senescence (SENS)
- Sub-lethal damage
- Transgenerational design
- Stem cell theory of aging
- Timeline of senescence research
References
- 1 2 Ainsworth, C; Lepage, M (2007). "Evolution's greatest mistakes". New Scientist. 195 (2616): 36–39. doi:10.1016/S0262-4079(07)62033-8.
- ↑ Walker, R.; Pakula, L.; Sutcliffe, M.; Kruk, P.; Graakjaer, J.; Shay, J. (2009). "A case study of "disorganized development" and its possible relevance to genetic determinants of aging". Mechanisms of ageing and development. 130 (5): 350–356. doi:10.1016/j.mad.2009.02.003. PMID 19428454.
- ↑ Brown, Bob (23 June 2006). "Doctors Baffled, Intrigued by Girl Who Doesn't Age". Health. ABC News. Retrieved 27 June 2009.
- ↑ Aubrey D.N.J, de Grey (2007). "Life Span Extension Research and Public Debate: Societal Considerations" (PDF). Studies in Ethics, Law, and Technology. 1 (1). doi:10.2202/1941-6008.1011. Article 5.
- ↑ "SENS Foundation".
- ↑ Hayflick L; Moorhead PS (December 1961). "The serial cultivation of human diploid cell strains". Exp. Cell Res. 25: 585–621. doi:10.1016/0014-4827(61)90192-6. PMID 13905658.
- ↑ Campisi, Judith (February 2013). "Aging, Cellular Senescence, and Cancer". Annual Review of Physiology. 75: 685–705. doi:10.1146/annurev-physiol-030212-183653. PMC 4166529
 . PMID 23140366.
. PMID 23140366. - ↑ Rodier, F.; Campisi, J. (14 February 2011). "Four faces of cellular senescence". The Journal of Cell Biology. 192 (4): 547–556. doi:10.1083/jcb.201009094.
- ↑ Burton, Dominick G. A.; Krizhanovsky, Valery (31 July 2014). "Physiological and pathological consequences of cellular senescence". Cellular and Molecular Life Sciences. 71 (22): 4373–4386. doi:10.1007/s00018-014-1691-3.
- 1 2 3 Baker, D.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.; Childs, B.; van de Sluis, B.; Kirkland, J.; van Deursen, J. (10 November 2011). "Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders". Nature. 479: 232–6. doi:10.1038/nature10600. PMC 3468323. PMID 22048312.
- ↑ Xu, M; Palmer, AK; Ding, H; Weivoda, MM; Pirtskhalava, T; White, TA; Sepe, A; Johnson, KO; Stout, MB; Giorgadze, N; Jensen, MD; LeBrasseur, NK; Tchkonia, T; Kirkland, JL (2015). "Targeting senescent cells enhances adipogenesis and metabolic function in old age". eLife. 4. doi:10.7554/eLife.12997. PMC 4758946. PMID 26687007.
- ↑ Quick, Darren (February 3, 2016). "Clearing out damaged cells in mice extends lifespan by up to 35 percent". www.gizmag.com. Retrieved 2016-02-04.
- ↑ Regalado, Antonio (February 3, 2016). "In New Anti-Aging Strategy, Clearing Out Old Cells Increases Life Span of Mice by 25 Percent". MIT Technology Review. Retrieved 2016-02-04.
- 1 2 Horvath S (2013). "DNA methylation age of human tissues and cell types". Genome Biology. 14: R115. doi:10.1186/gb-2013-14-10-r115. PMC 4015143. PMID 24138928.
- 1 2 3 Lowe, D (2016). "Epigenetic clock analyses of cellular senescence and ageing.". Oncotarget. 7 (8): 8524–8531. doi:10.18632/oncotarget.7383. PMID 26885756.
- ↑ "Aging and Gerontology Glossary". Retrieved 26 February 2011.
- ↑ Austad, S (2009). "Comparative Biology of Aging". J Gerontol a Biol Sci Med Sci. 64 (2): 199–201. doi:10.1093/gerona/gln060. PMC 2655036. PMID 19223603.
- ↑ Williams, G.C. (1957). "Pleiotropy, natural selection, and the evolution of senescence". Evolution. 11: 398–411. doi:10.2307/2406060.
- ↑ Emmanuel EJ. "Why I hope to die at 75: An argument that society and families – and you – will be better off if nature takes its course swiftly and promptly". The Atlantic. Retrieved 7 April 2015.
- ↑ Faria MA. "Bioethics and why I hope to live beyond age 75 attaining wisdom!: A rebuttal to Dr. Ezekiel Emanuel′s 75 age limit.". Surg Neurol Int 2015;6:35. Retrieved 7 April 2015.
- ↑ Faria MA. "Longevity and compression of morbidity from a neuroscience perspective: Do we have a duty to die by a certain age?". Surg Neurol Int 2015;6:49. Retrieved 7 April 2015.
- ↑ Thomas C. J. Tan; Ruman Rahman; Farah Jaber-Hijazi; Daniel A. Felix; Chen Chen; Edward J. Louis & Aziz Aboobaker (February 2012). "Telomere maintenance and telomerase activity are differentially regulated in asexual and sexual worms" (PDF). PNAS. 109 (9): 4209–4214. doi:10.1073/pnas.1118885109.
- ↑ Bowen RL; Atwood CS (2011). "The reproductive-cell cycle theory of aging: an update.". Experimental Gerontology. 46 (2): 100–7. doi:10.1016/j.exger.2010.09.007. PMID 20851172.
- ↑ Lopez-Otin, C; et al. (2013). "The hallmarks of aging.". Cell. 153: 1194–217. doi:10.1016/j.cell.2013.05.039. PMC 3836174. PMID 23746838.
- ↑ Medawar, P.B. (1952). An Unsolved problem of biology; an inaugural lecture delivered at University College, London, 6 December, 1951. London: H.K. Lewis. OCLC 8482093.
- ↑ Williams, G.C. (1957). "Pleiotropy, Natural Selection, and the Evolution of Senescence". Evolution. 11: 398–411. doi:10.2307/2406060.
- ↑ Hamilton WD (September 1966). "The moulding of senescence by natural selection". J. Theor. Biol. 12 (1): 12–45. doi:10.1016/0022-5193(66)90184-6. PMID 6015424.
- ↑ Kirkwood TB (November 1977). "Evolution of ageing". Nature. 270 (5635): 301–4. doi:10.1038/270301a0. PMID 593350.
- ↑ Fabian, Daniel; Flatt, Thomas (2011). "The Evolution of Aging". Scitable. Nature Publishing Group. Retrieved December 9, 2014.
- ↑ Medawar PB (1946). "Old age and natural death". Modern Quarterly. 1: 30–56.
- ↑ Medawar, Peter B. (1952). An Unsolved Problem of Biology. London: H. K. Lewis.
- ↑ Williams, George C. (December 1957). "Pleiotropy, Natural Selection, and the Evolution of Senescence" (PDF). Evolution. 11 (4): 398–411. doi:10.2307/2406060. JSTOR 2406060.
- 1 2 Bass TM; Weinkove D; Houthoofd K; Gems D; Partridge L (October 2007). "Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans". Mechanisms of Ageing and Development. 128 (10): 546–52. doi:10.1016/j.mad.2007.07.007. PMID 17875315.
- ↑ Ryley J; Pereira-Smith OM (2006). "Microfluidics device for single cell gene expression analysis in Saccharomyces cerevisiae". Yeast. 23 (14–15): 1065–73. doi:10.1002/yea.1412. PMID 17083143.
- ↑ "GenAge database". Retrieved 26 February 2011.
- ↑ Gavrilov LA; Gavrilova NS (December 2001). "The reliability theory of aging and longevity". Journal of Theoretical Biology. 213 (4): 527–45. doi:10.1006/jtbi.2001.2430. PMID 11742523.
- ↑ Stephens C (April 2005). "Senescence: even bacteria get old". Curr. Biol. 15 (8): R308–10. doi:10.1016/j.cub.2005.04.006. PMID 15854899.
- ↑ Burton; Faragher (2015). "Cellular senescence: from growth arrest to immunogenic conversion". AGE. 37. doi:10.1007/s11357-015-9764-2.
- ↑ Collins FS, et al. (13 June 2011). "Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts". J Clin Invest. 121 (7): 2833–44. doi:10.1172/JCI43578. PMC 3223819. PMID 21670498.
- ↑ Hanahan D; Weinberg RA (January 2000). "The hallmarks of cancer". Cell. 100 (1): 57–70. doi:10.1016/S0092-8674(00)81683-9. PMID 10647931.
- ↑ Krishnamurthy, J; Torrice, C; Ramsey, MR; Kovalev, GI; Al-Regaiey, K; Su, L; Sharpless, NE (2004). "Ink4a/Arf expression is a biomarker of aging". J. Clin. Invest. 114 (9): 1299–1307. doi:10.1172/JCI22475. PMC 524230. PMID 15520862.
- ↑ Baker DJ; Dawlaty MM; Wijshake T; Jeganathan KB; Malureanu L; van Ree JH; Crespo-Diaz R; Reyes S; Seaburg L; Shapiro V; Behfar A; Terzic A; van de Sluis B; van Deursen JM (Jan 2013). "Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan". Nat Cell Biol. 15 (1): 96–102. doi:10.1038/ncb2643. PMID 23242215.
- ↑ Zhu, Y; Tchkonia, T; Pirtskhalava, T; Gower, AC; Ding, H; Giorgadze, N; Palmer, AK; Ikeno, Y; Hubbard, GB; Lenburg, M; O'Hara, SP; LaRusso, NF; Miller, JD; Roos, CM; Verzosa, GC; LeBrasseur, NK; Wren, JD; Farr, JN; Khosla, S; Stout, MB; McGowan, SJ; Fuhrmann-Stroissnigg, H; Gurkar, AU; Zhao, J; Colangelo, D; Dorronsoro, A; Ling, YY; Barghouthy, AS; Navarro, DC; Sano, T; Robbins, PD; Niedernhofer, LJ; Kirkland, JL (9 March 2015). "The Achilles' heel of senescent cells: from transcriptome to senolytic drugs.". Aging Cell. 14: 644–58. doi:10.1111/acel.12344. PMID 25754370.
- ↑ Pearl, Raymond (1928). The Rate of Living, Being an Account of Some Experimental Studies on the Biology of Life Duration. New York: Alfred A. Knopf.
- ↑ Brunet-Rossinni AK; Austad SN (2004). "Ageing studies on bats: a review". Biogerontology. 5 (4): 211–22. doi:10.1023/B:BGEN.0000038022.65024.d8. PMID 15314271.
- ↑ de Magalhães JP; Costa J; Church GM (1 February 2007). "An Analysis of the Relationship Between Metabolism, Developmental Schedules, and Longevity Using Phylogenetic Independent Contrasts". The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 62 (2): 149–60. doi:10.1093/gerona/62.2.149. PMC 2288695. PMID 17339640. Archived from the original on 23 December 2014.
- ↑ Speakman JR; Selman C; McLaren JS; Harper EJ (1 June 2002). "Living fast, dying when? The link between aging and energetics". The Journal of Nutrition. 132 (6 Suppl 2): 1583S–97S. PMID 12042467.
- ↑ Freedman ND; Park Y; Abnet CC; Hollenbeck AR; Sinha R (May 2012). "Association of coffee drinking with total and cause-specific mortality". N. Engl. J. Med. 366 (20): 1891–904. doi:10.1056/NEJMoa1112010. PMC 3439152. PMID 22591295.
- ↑ Yang Y; Chan SW; Hu M; Walden R; Tomlinson B (2011). "Effects of some common food constituents on cardiovascular disease". ISRN Cardiol. 2011: 397136. doi:10.5402/2011/397136. PMC 3262529. PMID 22347642.
- ↑ Spencer RL; Hutchison KE (1999). "Alcohol, aging, and the stress response" (PDF). Alcohol Research & Health. 23 (4): 272–83. PMID 10890824.
- ↑ Alexander P (1967). "The role of DNA lesions in the processes leading to aging in mice". Symp. Soc. Exp. Biol. 21: 29–50. PMID 4860956.
- ↑ Gensler HL; Bernstein H (September 1981). "DNA damage as the primary cause of aging". Q Rev Biol. 56 (3): 279–303. doi:10.1086/412317. PMID 7031747.
- ↑ Bernstein C; Bernstein H (1991). Aging, Sex, and DNA Repair. San Diego CA: Academic Press. ISBN 0123960037.
- ↑ Ames BN; Gold LS (1991). "Endogenous mutagens and the causes of aging and cancer". Mutat. Res. 250 (1-2): 3–16. doi:10.1016/0027-5107(91)90157-j. PMID 1944345.
- ↑ Holmes GE; Bernstein C; Bernstein H (September 1992). "Oxidative and other DNA damages as the basis of aging: a review". Mutat. Res. 275 (3-6): 305–15. doi:10.1016/0921-8734(92)90034-M. PMID 1383772.
- ↑ Rao KS; Loeb LA (September 1992). "DNA damage and repair in brain: relationship to aging". Mutat. Res. 275 (3-6): 317–29. doi:10.1016/0921-8734(92)90035-N. PMID 1383773.
- ↑ Ames BN; Shigenaga MK; Hagen TM (September 1993). "Oxidants, antioxidants, and the degenerative diseases of aging". Proc. Natl. Acad. Sci. U.S.A. 90 (17): 7915–22. doi:10.1073/pnas.90.17.7915. PMC 47258. PMID 8367443.
- ↑ Bernstein, H; Payne, CM; Bernstein, C; Garewal, H; Dvorak, K (2008). "Cancer and aging as consequences of un-repaired DNA damage.". In Kimura, Honoka; Suzuki, Aoi. New Research on DNA Damage. Nova Science Publishers. pp. 1–47. ISBN 978-1604565812.
- ↑ Mounkes LC; Kozlov S (2003). "A progeroid syndrome in mice is caused by defects in A-type lamins" (PDF). Nature. 423 (6937): 298–301. doi:10.1038/nature01631.
- ↑ Macintosh, Kerry Lynn (2005). Illegal Beings: Human Clones and the Law. Cambridge: Cambridge University Press. ISBN 0-521-85328-1.
External links
| Look up senescence in Wiktionary, the free dictionary. |
- Senescence.info
- Fight Aging!
- SENS Foundation
- AgeLab (Massachusetts Institute of Technology)
- Senescence at DMOZ
- Aging Cell
- Telomere Shortening
- Jones, Owen R.; Scheuerlein, Alexander; Salguero-Gómez, Roberto; Camarda, Carlo Giovanni; Schaible, Ralf; Casper, Brenda B.; Dahlgren, Johan P.; Ehrlén, Johan; García, María B.; Menges, Eric S.; Quintana-Ascencio, Pedro F.; Caswell, Hal; Baudisch, Annette; Vaupel, James W. (2013). "Diversity of ageing across the tree of life". Nature. 505: 169–173. doi:10.1038/nature12789. Lay summary – National Geographic (8 December 2013).