Ca2+/calmodulin-dependent protein kinase II
| Calcium/calmodulin dependent protein kinase II association domain | |||||||||
|---|---|---|---|---|---|---|---|---|---|
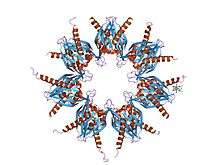 Crystal structure of calcium/calmodulin-dependent protein kinase | |||||||||
| Identifiers | |||||||||
| Symbol | CaMKII_AD | ||||||||
| Pfam | PF08332 | ||||||||
| Pfam clan | CL0051 | ||||||||
| InterPro | IPR013543 | ||||||||
| |||||||||
Ca2+
/calmodulin-dependent protein kinase II (CaM kinase II or CaMKII) is a serine/threonine-specific protein kinase that is regulated by the Ca2+
/calmodulin complex. CaMKII is involved in many signaling cascades and is thought to be an important mediator of learning and memory.[1]
CaMKII is also necessary for Ca2+
homeostasis and reuptake in cardiomyocytes,[2]
chloride transport in epithelia,[3]
positive T-cell selection,[4]
and CD8 T-cell activation.[5]
Misregulation of CaMKII is linked to Alzheimer’s disease, Angelman syndrome, and heart arrhythmia.[6]
Types
There are two types of CaM kinase:
- Specialized CaM kinases, such as the myosin light chain kinase that phosphorylates myosin, causing smooth muscles to contract
- Multifunctional CaM kinases, also collectively called CaM kinase II, which play a role in neurotransmitter secretion, transcription factor regulation, and glycogen metabolism.

Structure, function, and autoregulation

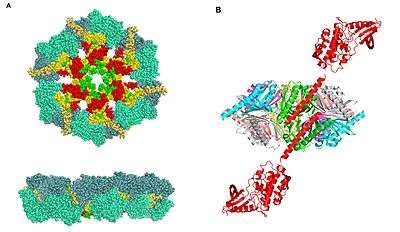
CaMKII accounts for 1–2% of all proteins in the brain, and has 28 different isoforms. The isoforms derive from the alpha, beta, gamma, and delta genes.
Structural domain
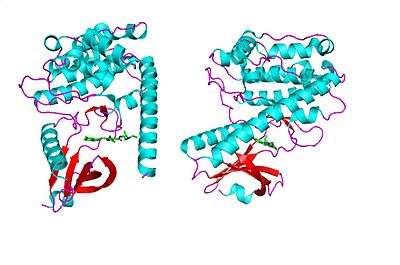
All of the isoforms of CaMKII have: a catalytic domain, an autoinhibitory domain, a variable segment, and a self-association domain.[7]
The catalytic domain has several binding sites for ATP and other substrate anchor proteins. It is responsible for the transfer of phosphate from ATP to Ser or Thr residues in substrates. The autoinhibitory domain features a pseudosubstrate site, which binds to the catalytic domain and blocks its ability to phosphorylate proteins.[8]
The structural feature that governs this autoinhibition is the Threonine 286 residue. Phosphorylation of this site will permanently activate the CaMKII enzyme. Once the Threonine 286 residue has been phosphorylated, the inhibitory domain is blocked from the pseudosubstrate site. This effectively blocks autoinhibition, allowing for permanent activation of the CaMKII enzyme. This enables CamKII to be active, even in the absence of calcium and calmodulin.[9]
The other two domains in CaMKII are the variable and self-association domains. Differences in these domains contribute to the various CaMKII isoforms.[10]
The self-association domain (CaMKII AD) is found at the C terminus, the function of this domain is the assembly of the single proteins into large (8 to 14 subunits) multimers [11]
Calcium and calmodulin dependence
The sensitivity of the CaMKII enzyme to calcium and calmodulin is governed by the variable and self-associative domains. This sensitivity level of CaMKII will also modulate the different states of activation for the enzyme. Initially, the enzyme is activated; however, autophosphorylation does not occur because there is not enough Calcium or calmodulin present to bind to neighboring subunits. As greater amounts of calcium and calmodulin accumulate, autophosphorylation occurs leading to persistent activation of the CaMKII enzyme for a short period of time. However, the Threonine 286 residue eventually becomes dephosphorylated, leading to inactivation of CaMKII.[12]
Autophosphorylation
Autophosphorylation is the process in which a kinase attaches a phosphate group to itself. When CaMKII autophosphorylates, it becomes persistently active. Phosphorylation of the Threonine 286 site allows for the activation of the catalytic domain. Autophosphorylation is enhanced by the structure of the holoenzyme because it is present in two stacked rings. The close proximity of these adjacent rings increases the probability of phosphorylation of neighboring CaMKII enzymes, furthering autophosphorylation.[13] A mechanism that promotes autophosphorylation features inhibition of the PP1 phosphatase. This enables CaMKII to be constantly active by increasing the likelihood of autophosphorylation.[14]
Long-term potentiation
Calcium/ calmodulin dependent protein kinase II is also heavily implicated in long-term potentiation (LTP) – the molecular process of strengthening active synapses that is thought to underlie the processes of memory. It is involved in many aspects of this process. LTP is initiated when the NMDA receptors (which act as "molecular coincidence receptors" and allow this process to be the result of BOTH pre- and post-synaptic neuron activation) allow Ca2+ into the post synaptic neuron. This Ca2+ influx activates CaMKII. It has been shown that there is an increase in CaMKII activity directly in the post synaptic density of dendrites after LTP induction, suggesting that activation is a direct result of stimulation.[15][16]
In LTP
When alpha-CaMKII is knocked out in mice, LTP is reduced by 50%. This can be explained by the fact that beta-CaMKII is responsible for approximately 65% of CaMKII activity.[17][18] LTP can be completely blocked if CaMKII is modified so that it cannot remain active.[2][19] After LTP induction, CaMKII moves to the postsynaptic density (PSD). However, if the stimulation does not induce LTP, the translocation is quickly reversible. Binding to the PSD changes CaMKII so that it is less likely to become dephosphorylated. CaMKII transforms from a substrate for Protein Phosphatase 2A (PP2A), which is responsible for dephosphorylating CaMKII, to that of Protein Phosphatase 1. Strack, S. (1997)[15] demonstrated this phenomenon by chemically stimulating hippocampal slices. This experiment illustrates that CaMKII contributes to the enhancement of synaptic strength. Sanhueza et al.[20] found that persistent activation of CaMKII is necessarily for the maintenance of LTP. She induced LTP in hippocampal slices and experimentally applied an antagonist (CaMKIINtide) to prevent CaMKII from remaining active. The slices that were applied with CaMKIINtide showed a decrease in Normalized EPSP slope after the drug infusion, meaning that the induced LTP reversed itself. The Normalized EPSP slope remained constant in the control; CaMKII continues to be involved in the LTP maintenance process even after LTP establishment. CaMKII is activated by calcium/calmodulin, but it is maintained by autophosphorylation. CaMKII is activated by the NMDA-receptor-mediated Calcium elevation that occurs during LTP induction. Activation is accompanied by phosphorylation of both the alpha and beta-subunits and Thr286/287.
Independent induction of LTP
LTP can be induced by artificially injecting CaMKII. When CaMKII is infused in postsynaptically in the hippocampal slices and intracellular perfusion or viral expression, there is a two- to threefold increase in the response of the synapse to glutamate and other chemical signals.[21][22]
Mechanistic role in LTP
There is strong evidence that after activation of CaMKII, CaMKII plays a role in the trafficking of AMPA receptors into the membrane and then the PSD of the dendrite. Movement of AMPA receptors increases postsynaptic response to presynaptic depolarization through strengthening the synapses. This produces LTP.
Mechanistically, CaMKII phosphorylates AMPA receptors at the P2 serine 831 site. This increases channel conductance of GluA1 subunits of AMPA receptors,[23] which allows AMPA receptors to be more sensitive than normal during LTP. Increased AMPA receptor sensitivity leads to increase synaptic strength.
In addition to increasing the channel conductance of GluA1 subunits, CaMKII has also been shown to aid in the process of AMPA receptor exocytosis. Reserve AMPA receptors are embedded in endosomes within the cell. CaMKII can stimulate the endosomes to move to the outer membrane and activate the embedded AMPA receptors.[24] Exocytosis of endosomes enlarges and increases the number of AMPA receptors in the synpase. The greater number of AMPA receptors increases the sensitivity of the synapse to presynaptic depolarization, and generates LTP.
Maintenance of LTP
Along with helping to establish LTP, CaMKII has been shown to be crucial in maintaining LTP. Its ability to autophosphorylate is thought to play an important role in this maintenance. Administration of certain CaMKII blockers has been shown not only to block LTP but also to reverse it in a time dependent manner.[25]
Behavioral Memory
As LTP is thought to underlie the processes of learning and memory, CaMKII is also crucial to memory formation. Behavioral studies involving genetically engineered mice have demonstrated the importance of CaMKII.
Preventing autophosphorylation
Deficit in spatial learning
In 1998, Giese and colleagues studied knockout mice that have been genetically engineered to prevent CaMKII autophosphorylation. They observed that mice had trouble finding the hidden platform in the Morris water maze task. The Morris water maze task is often used to represent hippocampus-dependent spatial learning. The mice’s inability to find the hidden platform implies deficits in spatial learning.[14]
However, these results were not entirely conclusive because memory formation deficit could also be associated with sensory motor impairment resulting from genetic alteration.[26]
Deficit in fear memories
Irvine and colleagues in 2006 showed that preventing autophosphorylation of CaMKII cause mice to have impaired initial learning of fear conditioning. However, after repeated trials, the impaired mice exhibited similar fear memory formation as the control mice. CaMKII may play a role in rapid fear memory, but does not completely prevent fear memory in the long run.[27]
In 2004, Rodrigues and colleagues found that fear conditioning increased phosphorylated CaMKII in lateral amygdala synapses and dendritic spines, indicating that fear conditioning could be responsible for regulating and activating the kinase. They also discovered a drug, KN-62, that inhibited CaMKII and prevented acquisition of fear conditioning and LTP.[28]
Deficit in consolidation of memory traces
α-CaMKII heterozygous mice express half the normal protein level as the wild-type level. These mice showed normal memory storage in the hippocampus, but deficits in consolidation of memory in the cortex.[29]
Overexpression
Mayford and colleagues engineered transgenic mice that express CaMKII with a point mutation of Thr-286 to aspartate, which mimics autophosphorylation and increases kinase activity. These mice failed to show LTP response to weak stimuli, and failed to perform hippocampus-dependent spatial learning that depended on visual or olfactory cues.[30]
Researchers speculate these results could be due to lack of stable hippocampal place cells in these animals.[31]
However, because genetic modifications might cause unintentional developmental changes, viral vector delivery allows the mice’s genetic material to be modified at specific stages of development. It is possible with viral vector delivery to inject a specific gene of choice into a particular region of the brain in an already developed animal. Poulsen and colleagues in 2007 used this method to inject CaMKII into the hippocampus. They found that overexpression of CaMKII resulted in slight enhancement of acquisition of new memories.[32]
Different forms
CaMK2A
CaMKIIA is one of the major forms of CamKII. It has been found to play a critical role in sustaining activation of CamKII at the postsynaptic density. Studies have found that knockout mice without CaMKIIA demonstrate a low frequency of LTP. Additionally, these mice do not form persistent, stable place cells in the hippocampus.[33]
CaMK2B
CaMK2B has an autophosphorylation site at Thr287. It functions as a targeting or docking module. Reverse transcription-polymerase chain reaction and sequencing analysis identified at least five alternative splicing variants of beta CaMKII (beta, beta6, betae, beta'e, and beta7) in brain and two of them (beta6 and beta7) were first detected in any species.[34]
CaMK2D
CaMK2D appears in both neuronal and non-neuronal cell types. It is characterized particularly in many tumor cells, such as a variety of pancreatic, leukemic, breast and other tumor cells.[35] found that CaMK2D is downregulated in human tumor cells.
CaMK2G
CaMK2G has been shown to be a crucial extracellular signal-regulated kinase in differentiated smooth muscle cells.[36]
Genes
References
- ↑ Yamauchi, Takashi (2005). "Neuronal Ca2+/Calmodulin-Dependent Protein Kinase II—Discovery, Progress in a Quarter of a Century, and Perspective: Implication for Learning and Memory". Biological & Pharmaceutical Bulletin. 28 (8): 1342–54. doi:10.1248/bpb.28.1342. PMID 16079472.
- 1 2 Anderson, M (2005). "Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias". Pharmacology & Therapeutics. 106: 39–55. doi:10.1016/j.pharmthera.2004.11.002.
- ↑ Fährmann, Michael; Kaufhold, Marc-André (2006). "Functional partitioning of epithelial protein kinase CaMKII in signal transduction". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1763: 101–9. doi:10.1016/j.bbamcr.2005.11.012.
- ↑ McGargill, Maureen A.; Sharp, Leslie L.; Bui, Jack D.; Hedrick, Stephen M.; Calbo, Sébastien (July 2005). "Active Ca2+
/calmodulin-dependent protein kinase II gamma B impairs positive selection of T cells by modulating TCR signaling". The Journal of Immunology. 175 (2): 656–64. doi:10.4049/jimmunol.175.2.656. PMID 16002660. - ↑ Lin, Meei Yun; Zal, Tomasz; Ch’en, Irene L.; Gascoigne, Nicholas R. J.; Hedrick, Stephen M. (May 2005). "A pivotal role for the multifunctional calcium/calmodulin-dependent protein kinase II in T cells: from activation to unresponsiveness". The Journal of Immunology. 174 (9): 5583–92. doi:10.4049/jimmunol.174.9.5583. PMID 15843557.
- ↑ Yamauchi, Takashi (August 2005). "Neuronal Ca2+
/calmodulin-dependent protein kinase II—discovery, progress in a quarter of a century, and perspective: implication for learning and memory". Biological & Pharmaceutical Bulletin. 28 (8): 1342–54. doi:10.1248/bpb.28.1342. PMID 16079472. - ↑ Hudmon, Andy; Schulman, Howard (2002). "Neuronal Ca2+/Calmodulin-Dependent Protein Kinase II: The Role of Structure and Autoregulation in Cellular Function". Annual Review of Biochemistry. 71: 473–510. doi:10.1146/annurev.biochem.71.110601.135410. PMID 12045104.
- ↑ Kanaseki, T; Ikeuchi, Y; Sugiura, H; Yamauchi, T (1991). "Structural features of Ca2+/calmodulin-dependent protein kinase II revealed by electron microscopy". The Journal of Cell Biology. 115 (4): 1049–60. doi:10.1083/jcb.115.4.1049. PMC 2289961
 . PMID 1659571.
. PMID 1659571. - ↑ Yang, E; Schulman, H (1999). "Structural examination of autoregulation of multifunctional calcium/calmodulin-dependent protein kinase II". The Journal of Biological Chemistry. 274 (37): 26199–208. doi:10.1074/jbc.274.37.26199. PMID 10473573.
- ↑ Giese, K. P. (1998). "Autophosphorylation at Thr286 of the Calcium-Calmodulin Kinase II in LTP and Learning". Science. 279 (5352): 26199–208. doi:10.1126/science.279.5352.870. PMID 9452388.
- ↑ Griffith LC, Lu CS, Sun XX (October 2003). "CaMKII, an enzyme on the move: regulation of temporospatial localization". Mol. Interv. 3 (7): 386–403. doi:10.1124/mi.3.7.386. PMID 14993460.
- ↑ Lisman, J (1994). "The CaM kinase II hypothesis for the storage of synaptic memory". Trends in Neurosciences. 17 (10): 406–12. doi:10.1016/0166-2236(94)90014-0. PMID 7530878.
- ↑ Blitzer, Robert D.; Wong, Tony; Nouranifar, Rabin; Iyengar, Ravi; Landau, Emmanuel M. (1995). "Postsynaptic CAMP pathway gates early LTP in hippocampal CA1 region". Neuron. 15 (6): 1403–14. doi:10.1016/0896-6273(95)90018-7. PMID 8845163.
- 1 2 Giese, K. P.; Fedorov, NB; Filipkowski, RK; Silva, AJ (1998). "Autophosphorylation at Thr286 of the Calcium-Calmodulin Kinase II in LTP and Learning". Science. 279 (5352): 870–3. doi:10.1126/science.279.5352.870. PMID 9452388.
- 1 2 Strack, S.; Choi, S; Lovinger, DM; Colbran, RJ (1997). "Translocation of Autophosphorylated Calcium/Calmodulin-dependent Protein Kinase II to the Postsynaptic Density". Journal of Biological Chemistry. 272 (21): 13467–70. doi:10.1074/jbc.272.21.13467. PMID 9153188.
- ↑ Gardoni, F; Schrama, LH; Kamal, A; Gispen, WH; Cattabeni, F; Di Luca, M (2001). "Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor". The Journal of neuroscience : the official journal of the Society for Neuroscience. 21 (5): 1501–9. PMID 11222640.
- ↑ Silva, A.; Stevens, C.; Tonegawa, S; Wang, Y (1992). "Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice". Science. 257 (5067): 201–6. doi:10.1126/science.1378648. PMID 1378648.
- ↑ Journal, pp. 344–54, doi:10.1101/lm.5.4.344
- ↑ Hrabetova, S; Sacktor, TC (1996). "Bidirectional regulation of protein kinase M zeta in the maintenance of long-term potentiation and long-term depression". The Journal of neuroscience : the official journal of the Society for Neuroscience. 16 (17): 5324–33. PMID 8757245.
- ↑ Sanhueza, M; McIntyre, CC; Lisman, JE (2007). "Reversal of synaptic memory by Ca2+/calmodulin-dependent protein kinase II inhibitor". The Journal of neuroscience : the official journal of the Society for Neuroscience. 27 (19): 5190–9. doi:10.1523/JNEUROSCI.5049-06.2007. PMID 17494705.
- ↑ Davies, SN; Lester, RA; Reymann, KG; Collingridge, GL (1989). "Temporally distinct pre- and post-synaptic mechanisms maintain long-term potentiation". Nature. 338 (6215): 500–3. doi:10.1038/338500a0. PMID 2564640.
- ↑ Montgomery, JM; Pavlidis, P; Madison, DV (2001). "Pair recordings reveal all-silent synaptic connections and the postsynaptic expression of long-term potentiation". Neuron. 29 (3): 691–701. doi:10.1016/S0896-6273(01)00244-6. PMID 11301028.
- ↑ Collingridge, Graham L.; Benke, Tim A.; Lüthi, Andreas; Isaac, John T. R. (1998). "Modulation of AMPA receptor unitary conductance by synaptic activity". Nature. 393 (6687): 793–7. doi:10.1038/31709. PMID 9655394.
- ↑ Lisman, John; Schulman, Howard; Cline, Hollis (2002). "The Molecular Basis of CaMKII Function in Synaptic and Behavioural Memory". Nature Reviews Neuroscience. 3 (3): 175–90. doi:10.1038/nrn753. PMID 11994750.
- ↑ Yang, H.-W.; Hu, XD; Zhang, HM; Xin, WJ; Li, MT; Zhang, T; Zhou, LJ; Liu, XG (2003). "Roles of CaMKII, PKA, and PKC in the Induction and Maintenance of LTP of C-Fiber-Evoked Field Potentials in Rat Spinal Dorsal Horn". Journal of Neurophysiology. 91 (3): 1122–33. doi:10.1152/jn.00735.2003. PMID 14586032.
- ↑ Rudy, Jerry W. (2004). The Neurobiology of Learning and Memory. Snauer. ISBN 978-0-87893-669-4.
- ↑ Irvine, Elaine E.; Von Hertzen, Laura S. J.; Plattner, Florian; Giese, Karl Peter (2006). "αCaMKII autophosphorylation: a fast track to memory". Trends in Neurosciences. 29 (8): 459–65. doi:10.1016/j.tins.2006.06.009. PMID 16806507.
- ↑ Rodrigues, S. M.; Farb, CR; Bauer, EP; Ledoux, JE; Schafe, GE (2004). "Pavlovian Fear Conditioning Regulates Thr286 Autophosphorylation of Ca2+/Calmodulin-Dependent Protein Kinase II at Lateral Amygdala Synapses". Journal of Neuroscience. 24 (13): 3281–8. doi:10.1523/JNEUROSCI.5303-03.2004. PMID 15056707.
- ↑ Frankland, Paul W.; O'Brien, Cara; Ohno, Masuo; Kirkwood, Alfredo; Silva, Alcino J. (2001). "Alpha-CaMKII-dependent plasticity in the cortex is required for permanent memory". Nature. 411 (6835): 309–13. doi:10.1038/35077089. PMID 11357133.
- ↑ Mayford, Mark; Wang, Jian; Kandel, Eric R; O'Dell, Thomas J (1995). "CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP". Cell. 81 (6): 891–904. doi:10.1016/0092-8674(95)90009-8. PMID 7781066.
- ↑ Rotenberg, Alexander; Mayford, Mark; Hawkins, Robert D; Kandel, Eric R; Muller, Robert U (1996). "Mice Expressing Activated CaMKII Lack Low Frequency LTP and Do Not Form Stable Place Cells in the CA1 Region of the Hippocampus". Cell. 87 (7): 1351–61. doi:10.1016/S0092-8674(00)81829-2. PMID 8980240.
- ↑ Poulsen, D.J.; Standing, D.; Bullshields, K.; Spencer, K.; Micevych, P.E.; Babcock, A.M. (2007). "Overexpression of hippocampal Ca2+/calmodulin-dependent protein kinase II improves spatial memory". Journal of Neuroscience Research. 85 (4): 735–9. doi:10.1002/jnr.21163. PMID 17171706.
- ↑ Soderling, T (2000). "CaM-kinases: modulators of synaptic plasticity". Current Opinion in Neurobiology. 10 (3): 375–80. doi:10.1016/S0959-4388(00)00090-8. PMID 10851169.
- ↑ Wang, P; Wu, YL; Zhou, TH; Sun, Y; Pei, G (2000). "Identification of alternative splicing variants of the β subunit of human Ca2+/calmodulin-dependent protein kinase II with different activities". FEBS Letters. 475 (2): 107–10. doi:10.1016/S0014-5793(00)01634-3. PMID 10858498.
- ↑ Wang, P; Wu, YL; Zhou, TH; Sun, Y; Pei, G (2000). "Identification of alternative splicing variants of the β subunit of human Ca2+/calmodulin-dependent protein kinase II with different activities". FEBS Letters. 475 (2): 1–11. doi:10.1016/S0014-5793(00)01634-3. PMID 10858498.
- ↑ Marganski, W. A.; Gangopadhyay, SS; Je, HD; Gallant, C; Morgan, KG (2005). "Targeting of a Novel Ca+2/Calmodulin-Dependent Protein Kinase II Is Essential for Extracellular Signal-Regulated Kinase-Mediated Signaling in Differentiated Smooth Muscle Cells". Circulation Research. 97 (6): 541–549. doi:10.1161/01.RES.0000182630.29093.0d. PMID 16109919.
External links
- Calcium-Calmodulin Dependent Protein Kinases at the US National Library of Medicine Medical Subject Headings (MeSH)
- To learn more about the CaMKII ...
This article incorporates text from the public domain Pfam and InterPro IPR013543