Chemical biology
Chemical biology is a scientific discipline spanning the fields of chemistry, biology, and physics. It involves the application of chemical techniques, tools, and analyses, and often compounds produced through synthetic chemistry, to the study and manipulation of biological systems. Chemical biologists attempt to use chemical principles to modulate systems to either investigate the underlying biology or create new function. Research done by chemical biologists is often closer related to that of cell biology than biochemistry. Biochemists study the chemistry of biomolecules and regulation of biochemical pathways within cells and tissues, e.g. cAMP or cGMP, while chemical biologists deal with novel chemical compounds applied to biology.
Introduction
Some forms of chemical biology attempt to answer biological questions by directly probing living systems at the chemical level. In contrast to research using biochemistry, genetics, or molecular biology, where mutagenesis can provide a new version of the organism or cell of interest, chemical biology studies probe systems in vitro and in vivo with small molecules that have been designed for a specific purpose or identified on the basis of biochemical or cell-based screening.
Chemical biology is one of many interfacial sciences that are characteristic of a general trend away from older, reductionist fields toward those whose goals are to achieve a description of scientific holism. In this sense, it is related to other fields such as proteomics. Chemical biology has scientific, historical and philosophical roots in medicinal chemistry, supramolecular chemistry (particularly host-guest chemistry), bioorganic chemistry, pharmacology, genetics, biochemistry, and metabolic engineering.
Systems of interest
Proteomics
Proteomics investigates the proteome, the set of expressed proteins at a given time under defined conditions. As a discipline, proteomics has moved past rapid protein identification and has developed into a biological assay for quantitative analysis of complex protein samples by comparing protein changes in differently perturbed systems.[1] Current goals in proteomics include determining protein sequences, abundance and any post-translational modifications. Also of interest are protein–protein interactions, cellular distribution of proteins and understanding protein activity. Another important aspect of proteomics is the advancement of technology to achieve these goals.
Protein levels, modifications, locations, and interactions are complex and dynamic properties. With this complexity in mind, experiments need to be carefully designed to answer specific questions especially in the face of the massive amounts of data that are generated by these analyses. The most valuable information comes from proteins that are expressed differently in a system being studied. These proteins can be compared relative to each other using quantitative proteomics, which allows a protein to be labeled with a mass tag. Proteomic technologies must be sensitive and robust, it is for these reasons, the mass spectrometer has been the workhorse of protein analysis. The high precision of mass spectrometry can distinguish between closely related species and species of interest can be isolated and fragmented within the instrument. Its applications to protein analysis was only possible in the late 1980s with the development of protein and peptide ionization with minimal fragmentation. These breakthroughs were ESI and MALDI. Mass spectrometry technologies are modular and can be chosen or optimized to the system of interest.
Chemical biologists are poised to impact proteomics through the development of techniques, probes and assays with synthetic chemistry for the characterization of protein samples of high complexity. These approaches include the development of enrichment strategies, chemical affinity tags and probes.
Enrichment techniques
Samples for Proteomics contain a myriad of peptide sequences, the sequence of interest may be highly represented or of low abundance. However, for successful MS analysis the peptide should be enriched within the sample. Reduction of sample complexity is achieved through selective enrichment using affinity chromatography techniques. This involves targeting a peptide with a distinguishing feature like a biotin label or a post translational modification.[2] Interesting methods have been developed that include the use of antibodies, lectins to capture glycoproteins, immobilized metal ions to capture phosphorylated peptides and suicide enzyme substrates to capture specific enzymes. Here, chemical biologists can develop reagents to interact with substrates, specifically and tightly, to profile a targeted functional group on a proteome scale. Development of new enrichment strategies is needed in areas like non-ser/thr/tyr phosphorylation sites and other post translational modifications. Other methods of decomplexing samples relies on upstream chromatographic separations.
Affinity tags
Chemical synthesis of affinity tags has been crucial to the maturation of quantitative proteomics. iTRAQ, Tandem mass tags (TMT) and Isotope-coded affinity tag (ICAT) are protein mass-tags that consist of a covalently attaching group, a mass (isobaric or isotopic) encoded linker and a handle for isolation. Varying mass-tags bind to different proteins as a sort of footprint such that when analyzing cells of differing perturbations, the levels of each protein can be compared relatively after enrichment by the introduced handle. Other methods include SILAC and heavy isotope labeling. These methods have been adapted to identify complexing proteins by labeling a bait protein, pulling it down and analyzing the proteins it has complexed.[3] Another method creates an internal tag by introducing novel amino acids that are genetically encoded in prokaryotic and eukaryotic organisms. These modifications create a new level of control and can facilitate photocrosslinking to probe protein–protein interactions.[4] In addition, keto, acetylene, azide, thioester, boronate, and dehydroalanine- containing amino acids can be used to selectively introduce tags, and novel chemical functional groups into proteins.[5]
Enzyme probes
To investigate enzymatic activity as opposed to total protein, activity-based reagents have been developed to label the enzymatically active form of proteins (see Activity-based proteomics). For example, serine hydrolase- and cysteine protease-inhibitors have been converted to suicide inhibitors.[6] This strategy enhances the ability to selectively analyze low abundance constituents through direct targeting. Structures that mimic these inhibitors could be introduced with modifications that will aid proteomic analysis- like an identification handle or mass tag.[7] Enzyme activity can also be monitored through converted substrate.[8] This strategy relies on using synthetic substrate conjugates that contain moieties that are acted upon by specific enzymes. The product conjugates are then captured by an affinity reagent and analyzed. The measured concentration of product conjugate allow the determination of the enzyme velocity. Identification of enzyme substrates (of which there may be hundreds or thousands, many of which unknown) is a problem of significant difficulty in proteomics and is vital to the understanding of signal transduction pathways in cells; techniques for labelling cellular substrates of enzymes is an area chemical biologists can address. A method that has been developed uses "analog-sensitive" kinases to label substrates using an unnatural ATP analog, facilitating visualization and identification through a unique handle.[9]
Glycobiology
While DNA, RNA and proteins are all encoded at the genetic level, there exists a separate system of trafficked molecules in the cell that are not encoded directly at any direct level: sugars. Thus, glycobiology is an area of dense research for chemical biologists. For instance, live cells can be supplied with synthetic variants of natural sugars in order to probe the function of the sugars in vivo. Carolyn Bertozzi at University of California, Berkeley has developed a method for site-specifically reacting molecules the surface of cells that have been labeled with synthetic sugars.
Combinatorial chemistry
Chemical biologists used automated synthesis of many diverse compounds in order to experiment with effects of small molecules on biological processes. More specifically, they observe changes in the behaviors of proteins when small molecules bind to them. Such experiments may supposedly lead to discovery of small molecules with antibiotic or chemotherapeutic properties. These approaches are identical to those employed in the discipline of pharmacology.
Molecular sensing
Chemical biologists are also interested in developing new small-molecule and biomolecule-based tools to study biological processes, often by molecular imaging techniques.[10] The field of molecular sensing was popularized by Roger Tsien's work developing calcium-sensing fluorescent compounds as well as pioneering the use of GFP, for which he was awarded the 2008 Nobel Prize in Chemistry.[11] Today, researchers continue to utilize basic chemical principles to develop new compounds for the study of biological metabolites and processes.
siRNA-A tool in chemical biology
siRNA or small interfering RNAs owe their origins to the difficulties the scientific community faced utilizing classical and reverse genetics methods in studying gene expression. Disrupting genes to study their functions is not always optimal; neither is mapping mutations back to their genes easy. The whole process is expensive as well as time-consuming, which is why a lot of effort has been devoted to develop methods to silence gene expression in sequence specific manner using nucleic acids. They have the potential to be powerful tools in the field of chemical biology to study the chemistry of gene expression in therapeutic targets of bacteria and viruses.
A number of different types of nucleic acid molecules have already gained prominence because of their potential as therapeutics. They target mRNAs to silence the genes in a sequence specific manner. Oligodeoxyribonucleic acids, ODNs[12] utilize steric interaction to silence gene expression. They can also form triple helices in conjunction with the DNA duplex. Whereas ribozymes[13][14] can be chemically designed to target specific genes and cleave them in a sequence specific manner. The most promising of these methods however is utilization of short interfering RNA or siRNA to silence gene expression.
siRNA
siRNA or short interfering RNAs exist in nature as a means for the express purpose of controlling gene expression. It was discovered in petunia as a post-transcriptional gene silencing measure.[15] It is the resultant product when a long double-strand RNA of 20 -25 nucleotides length was processed in the cells by the enzyme DICER. The newly synthesized siRNA assemble into endoribonuclease-containing complexes known as RNA-induced silencing complexes (RISCs), unwinding in the process. The activated RISC then binds to the complementary RNA molecules by base pairing interactions between the siRNA strand and the mRNA, which is then cleaved. This mechanism is known as RNA interference or RNAi.[16][17][18]
Designing and synthesizing siRNAs
It is now possible to order siRNAs designed and synthesized with the express purpose of targeting a particular sequence. The ambion website has a lot of information on the optimal design of siRNAs.
siRNAs can be synthesized chemically, or enzymatically. RNase III or DICER can be used to cleave the long dsRNAs to produce siRNAs. However the most expedient method is the use of plasmids to express them in vivo by delivering them into the target cell using vectors. This method allows the siRNAs to be expressed in the target cell stably, over a period of time and overcomes the drawbacks of the transience of their effect. Numerous strategies have been developed in order to deliver the siRNA into the cell efficiently:[19]
- Electroporation
- Local and systemic injection: This method was the first success scientists had in silencing genes using siRNAs. They were successfully delivered into highly vascularized tissue in mice through using high-pressure tail vein injection. Greater than 90% loss in gene expression was observed in the targets.
- siRNA producing viruses: This method shows great promise in gene therapy, and research is progressing in order to generate recombinant viruses that can produce siRNA in target cells.
- Small molecules that enhance transdermal penetration: Research in this field is moving at a fast pace in order to synthesize small organic molecules that, if injected in conjunction with siRNAs, can help them penetrate into the target cells.
Biological uses of the RNAi approach
The principal purpose of studying siRNA mediated RNA interference is probably to investigate gene function.[20]
It is so much easier to make genetic knock-outs by simply introducing sequence-specific siRNAs into cells; multi-copy genes can be silenced in one fell swoop by this method. Creation of double-knockout mutants is also easier and consumes much less time. Using local injections in specific regions of the model organisms also help in creating spatially separated and restricted knockout. siRNAs are also being successfully used to screen whole genomes in organisms such as C. elegans and Drosophilla melanogaster. Even in mammalian systems such as Danio rerio (zebrafish) that usually prove intractable to all gene silencing methods, even dsRNA injection, siRNA can do the job. It is paving a new way in development of therapeutics by identifying human gene orthologs in other species in a remarkably short period of time.
Numerous high-throughput screening approaches are being developed to screen large libraries of cells rapidly in order to identify drug targets.[21] A brief description of few of the screening techniques:
- Pooled Format Screening: A reagent library of RNAi has to be introduced to the cells so that a particular cell is in one particular reagent. The primary hits are then identified and their identity elucidate by sequencing techniques.
- Arrayed Format Screening: Each RNAi reagent is placed in separate wells in a plate and multiple manipulations can be done to identify their targets, which are then detected by fluorescence readouts, imaging techniques and other methods as well. Thus the identity of the target cell can be determined through the identity of the reagent in the database.
- Multiplexed methods: A combination of various assays can be used for high-throughput screening of candidate drug targets. For example, candidate genes can be identified through informatics based methods and then screened against a library of reagents. Many other such methods are being developed in order to make the job of screening therapeutic targets easier.
siRNA based therapeutics
The future in this field rests in the development of siRNA-based drugs.[22]
This could prove to be a powerful tool in gene based therapy. Research is now concentrated on developing strategies to design siRNA therapeutics for clinical use. A brief description of some novel strategies for siRNA drug development is provided here:
- Direct Mutation Targeting: The siRNAs are designed to perfectly match mutant alleles but contain one or more mismatches with wild-type alleles, leading to specific degradation of the matching, mutant transcripts.
- Indirect Mutation Targeting: The siRNA approach will not work if the mutant alleles are too similar to wild type. So an indirect approach is taken in which siRNAs are designed against disease linked markers such as SNP variations. The ones that are screened as positive are targeted for degradation.
- Exon-specific targeting: siRNAs are designed to target expressed regions (exons) of the gene.
- Targeting exon skipped transcripts: If the problem in the gene lies in aberrant splicing post-transcription, siRNA can be designed to target the unnatural exon-exon interface arising as a result of such alternative splicing.
Employing biology
Many research programs are also focused on employing natural biomolecules to perform a task or act as support for a new chemical method or material. In this regard, researchers have shown that DNA can serve as a template for synthetic chemistry, self-assembling proteins can serve as a structural scaffold for new materials, and RNA can be evolved in vitro to produce new catalytic function.
Protein misfolding and aggregation as a cause of disease
A common form of aggregation is long, ordered spindles called amyloid fibrils that are implicated in Alzheimer’s disease and that have been shown to consist of cross-linked beta sheet regions perpendicular to the backbone of the polypeptide.[23] Another form of aggregation occurs with prion proteins, the glycoproteins found with Creutzfeldt–Jakob disease and bovine spongiform encephalopathy. In both structures, aggregation occurs through hydrophobic interactions and water must be excluded from the binding surface before aggregation can occur.[24] A movie of this process can be seen in "Chemical and Engineering News".[25] The diseases associated with misfolded proteins are life-threatening and extremely debilitating, which makes them an important target for chemical biology research.
Through the transcription and translation process, DNA encodes for specific sequences of amino acids. The resulting polypeptides fold into more complex secondary, tertiary, and quaternary structures to form proteins. Based on both the sequence and the structure, a particular protein is conferred its cellular function. However, sometimes the folding process fails due to mutations in the genetic code and thus the amino acid sequence or due to changes in the cell environment (e.g. pH, temperature, reduction potential, etc.). Misfolding occurs more often in aged individuals or in cells exposed to a high degree of oxidative stress, but a fraction of all proteins misfold at some point even in the healthiest of cells.
Normally when a protein does not fold correctly, molecular chaperones in the cell can encourage refolding back into its active form. When refolding is not an option, the cell can also target the protein for degradation back into its component amino acids via proteolytic, lysosomal, or autophagic mechanisms. However, under certain conditions or with certain mutations, the cells can no longer cope with the misfolded protein(s) and a disease state results. Either the protein has a loss-of-function, such as in cystic fibrosis, in which it loses activity or cannot reach its target, or the protein has a gain-of-function, such as with Alzheimer's disease, in which the protein begins to aggregate causing it to become insoluble and non-functional.
Protein misfolding has previously been studied using both computational approaches as well as in vivo biological assays in model organisms such as Drosophila melanogaster and C. elegans. Computational models use a de novo process to calculate possible protein structures based on input parameters such as amino acid sequence, solvent effects, and mutations. This method has the shortcoming that the cell environment has been drastically simplified, which limits the factors that influence folding and stability. On the other hand, biological assays can be quite complicated to perform in vivo with high-throughput like efficiency and there always remains the question of how well lower organism systems approximate human systems.
Dobson et al. propose combining these two approaches such that computational models based on the organism studies can begin to predict what factors will lead to protein misfolding.[26] Several experiments have already been performed based on this strategy. In experiments on Drosophila, different mutations of beta amyloid peptides were evaluated based on the survival rates of the flies as well as their motile ability. The findings from the study show that the more a protein aggregates, the more detrimental the neurological dysfunction.[26][27][28] Further studies using transthyretin, a component of cerebrospinal fluid that binds to beta amyloid peptide deterring aggregation but can itself aggregate especially when mutated, indicate that aggregation prone proteins may not aggregate where they are secreted and rather are deposited in specific organs or tissues based on each mutation.[29] Kelly et al. have shown that the more stable, both kinetically and thermodynamically, a misfolded protein is the more likely the cell is to secrete it from the endoplasmic reticulum rather than targeting the protein for degradation.[30] In addition, the more stress that a cell feels from misfolded proteins the more probable new proteins will misfold.[31] These experiments as well as others having begun to elucidate both the intrinsic and extrinsic causes of misfolding as well as how the cell recognizes if proteins have folded correctly.
As more information is obtained on how the cell copes with misfolded proteins, new therapeutic strategies begin to emerge. An obvious path would be prevention of misfolding. However, if protein misfolding cannot be avoided, perhaps the cell's natural mechanisms for degradation can be bolstered to better deal with the proteins before they begin to aggregate.[32] Before these ideas can be realized, many more experiments need to be done to understand the folding and degradation machinery as well as what factors lead to misfolding. More information about protein misfolding and how it relates to disease can be found in the recently published book by Dobson, Kelly, and Rameriz-Alvarado entitled Protein Misfolding Diseases Current and Emerging Principles and Therapies.[33]
Chemical synthesis of peptides
In contrast to the traditional biotechnological practice of obtaining peptides or proteins by isolation from cellular hosts through cellular protein production, advances in chemical techniques for the synthesis and ligation of peptides has allowed for the total synthesis of some peptides and proteins. Chemical synthesis of proteins is a valuable tool in chemical biology as it allows for the introduction of non-natural amino acids as well as residue specific incorporation of "posttranslational modifications" such as phosphorylation, glycosylation, acetylation, and even ubiquitination. These capabilities are valuable for chemical biologists as non-natural amino acids can be used to probe and alter the functionality of proteins, while post translational modifications are widely known to regulate the structure and activity of proteins. Although strictly biological techniques have been developed to achieve these ends, the chemical synthesis of peptides often has a lower technical and practical barrier to obtaining small amounts of the desired protein. Given the widely recognized importance of proteins as cellular catalysts and recognition elements, the ability to precisely control the composition and connectivity of polypeptides is a valued tool in the chemical biology community and is an area of active research.
While chemists have been making peptides for over 100 years,[34] the ability to efficiently and quickly synthesize short peptides came of age with the development of Bruce Merrifield's solid phase peptide synthesis (SPPS). Prior to the development of SPPS, the concept of step-by-step polymer synthesis on an insoluble support was without chemical precedent.[35] The use of a covalently bound insoluble polymeric support greatly simplified the process of peptide synthesis by reducing purification to a simple "filtration and wash" procedure and facilitated a boom in the field of peptide chemistry. The development and "optimization" of SPPS took peptide synthesis from the hands of the specialized peptide synthesis community and put it into the hands of the broader chemistry, biochemistry, and now chemical biology community. SPPS is still the method of choice for linear synthesis of polypeptides up to 50 residues in length[35] and has been implemented in commercially available automated peptide synthesizers. One inherent shortcoming in any procedure that calls for repeated coupling reactions is the buildup of side products resulting from incomplete couplings and side reactions. This places the upper bound for the synthesis of linear polypeptide lengths at around 50 amino acids, while the "average" protein consists of 250 amino acids.[34] Clearly, there was a need for development of "non-linear" methods to allow synthetic access to the average protein.
Although the shortcomings of linear SPPS were recognized not long after its inception, it took until the early 1990s for effective methodology to be developed to ligate small peptide fragments made by SPPS, into protein sized polypeptide chains (for recent review of peptide ligation strategies, see review by Dawson et al.[36]). The oldest and best developed of these methods is termed native chemical ligation. Native chemical ligation was unveiled in a 1994 paper from the laboratory of Stephen B. H. Kent.[37] Native chemical ligation involves the coupling of a C-terminal thioester and an N-terminal cysteine residue, ultimately resulting in formation of a "native" amide bond. Further refinements in native chemical ligation have allowed for kinetically controlled coupling of multiple peptide fragments, allowing access to moderately sized peptides such as an HIV-protease dimer[38] and human lysozyme.[39] Even with the successes and attractive features of native chemical ligation, there are still some drawbacks in the utilization of this technique. Some of these drawbacks include the installation and preservation of a reactive C-terminal thioester, the requirement of an N-terminal cysteine residue (which is the second-least-common amino acid in proteins),[40] and the requirement for a sterically unincumbering C-terminal residue.
Other strategies that have been used for the ligation of peptide fragments using the acyl transfer chemistry first introduced with native chemical ligation include expressed protein ligation,[41] sulfurization/desulfurization techniques,[42] and use of removable thiol auxiliaries.[43]
Expressed protein ligation allows for the biotechnological installation of a C-terminal thioester using intein biochemistry, thereby allowing the appendage of a synthetic N-terminal peptide to the recombinantly produced C-terminal portion. This technique allows for access to much larger proteins, as only the N-terminal portion of the resulting protein has to be chemically synthesized. Both sulfurization/desulfurization techniques and the use of removable thiol auxiliaries involve the installation of a synthetic thiol moiety to carry out the standard native chemical ligation chemistry, followed by removal of the auxiliary/thiol. These techniques help to overcome the requirement of an N-terminal cysteine needed for standard native chemical ligation, although the steric requirements for the C-terminal residue are still limiting.
A final category of peptide ligation strategies include those methods not based on native chemical ligation type chemistry. Methods that fall in this category include the traceless Staudinger ligation,[44] azide-alkyne dipolar cycloadditions,[45] and imine ligations.[46]
Major contributors in this field today include Stephen B. H. Kent, Philip E. Dawson, and Tom W. Muir, as well as many others involved in methodology development and applications of these strategies to biological problems.
Protein design by directed evolution
One of the primary goals of protein engineering is the design of novel peptides or proteins with a desired structure and chemical activity. Because our knowledge of the relationship between primary sequence, structure, and function of proteins is limited, rational design of new proteins with enzymatic activity is extremely challenging. Directed evolution, repeated cycles of genetic diversification followed by a screening or selection process, can be used to mimic Darwinian evolution in the laboratory to design new proteins with a desired activity.[47]
Several methods exist for creating large libraries of sequence variants. Among the most widely used are subjecting DNA to UV radiation or chemical mutagens, error-prone PCR, degenerate codons, or recombination.[48][49] Once a large library of variants is created, selection or screening techniques are used to find mutants with a desired attribute. Common selection/screening techniques include fluorescence-activated cell sorting (FACS),[50] mRNA display,[51] phage display, or in vitro compartmentalization.[52] Once useful variants are found, their DNA sequence is amplified and subjected to further rounds of diversification and selection. Since only proteins with the desired activity are selected, multiple rounds of directed evolution lead to proteins with an accumulation beneficial traits.
There are two general strategies for choosing the starting sequence for a directed evolution experiment: de novo design and redesign. In a protein design experiment, an initial sequence is chosen at random and subjected to multiple rounds of directed evolution. For example, this has been employed successfully to create a family of ATP-binding proteins with a new folding pattern not found in nature.[53] Random sequences can also be biased towards specific folds by specifying the characteristics (such as polar vs. nonpolar) but not the specific identity of each amino acid in a sequence. Among other things, this strategy has been used to successfully design four-helix bundle proteins.[54][55] Because it is often thought that a well-defined structure is required for activity, biasing a designed protein towards adopting a specific folded structure is likely to increase the frequency of desirable variants in constructed libraries.
In a protein redesign experiment, an existing sequence serves as the starting point for directed evolution. In this way, old proteins can be redesigned for increased activity or new functions. Protein redesign has been used for protein simplification, creation of new quaternary structures, and topological redesign of a chorismate mutase.[48][56][57] To develop enzymes with new activities, one can take advantage of promiscuous enzymes or enzymes with significant side reactions. In this regard, directed evolution has been used on γ-humulene synthase, an enzyme that creates over 50 different sesquiterpenes, to create enzymes that selectively synthesize individual products.[58] Similarly, completely new functions can be selected for from existing protein scaffolds. In one example of this, an RNA ligase was created from a zinc finger scaffold after 17 rounds of directed evolution. This new enzyme catalyzes a chemical reaction not known to be catalyzed by any natural enzyme.[59]
Computational methods, when combined with experimental approaches, can significantly assist both the design and redesign of new proteins through directed evolution. Computation has been used to design proteins with unnatural folds, such as a right-handed coiled coil.[60] These computational approaches could also be used to redesign proteins to selectively bind specific target molecules. By identifying lead sequences using computational methods, the occurrence of functional proteins in libraries can be dramatically increased before any directed evolution experiments in the laboratory.
Manfred T. Reetz, Frances Arnold, Donald Hilvert, and Jack W. Szostak are significant researchers in this field.
Biocompatible click cycloaddition reactions in chemical biology
Recent advances in technology have allowed scientists to view substructures of cells at levels of unprecedented detail. Unfortunately these "aerial" pictures offer little information about the mechanics of the biological system in question. To be fully effective, precise imaging systems require a complementary technique that better elucidates the machinery of a cell. By attaching tracking devices (optical probes) to biomolecules in vivo, one can learn far more about cell metabolism, molecular transport, cell-cell interactions and many other processes[61]
Bioorthogonal reactions
Successful labeling of a molecule of interest requires specific functionalization of that molecule to react chemospecifically with an optical probe. For a labeling experiment to be considered robust, that functionalization must minimally perturb the system.[62] Unfortunately, these requirements can often be extremely hard to meet. Many of the reactions normally available to organic chemists in the laboratory are unavailable in living systems. Water- and redox- sensitive reactions would not proceed, reagents prone to nucleophilic attack would offer no chemospecificity, and any reactions with large kinetic barriers would not find enough energy in the relatively low-heat environment of a living cell. Thus, chemists have recently developed a panel of bioorthogonal chemistry that proceed chemospecifically, despite the milieu of distracting reactive materials in vivo.
Design of bioorthogonal reagents and bioorthogonal chemical reporters
The coupling of an optical probe to a molecule of interest must occur within a reasonably short time frame; therefore, the kinetics of the coupling reaction should be highly favorable. Click chemistry is well suited to fill this niche, since click reactions are, by definition, rapid, spontaneous, selective, and high-yielding.[63] Unfortunately, the most famous "click reaction," a [3+2] cycloaddition between an azide and an acyclic alkyne, is copper-catalyzed, posing a serious problem for use in vivo due to copper's toxicity.[64]
The issue of copper toxicity can be alleviated using copper-chelating ligands, enabling copper-catalyzed labeling of the surface of live cells.[65]
To bypass the necessity for a catalyst, the lab of Dr. Carolyn Bertozzi introduced inherent strain into the alkyne species by using a cyclic alkyne. In particular, cyclooctyne reacts with azido-molecules with distinctive vigor.[66] Further optimization of the reaction led to the use of difluorinated cyclooctynes (DIFOs), which increased yield and reaction rate.[67] Other coupling partners discovered by separate labs to be analogous to cyclooctynes include trans cyclooctene,[68] norbornene,[69] and a cyclobutene-functionalized molecule.[70]
Use in biological systems
As mentioned above, the use of bioorthogonal reactions to tag biomolecules requires that one half of the reactive "click" pair is installed in the target molecule, while the other is attached to an optical probe. When the probe is added to a biological system, it will selectively conjugate with the target molecule.
The most common method of installing bioorthogonal reactivity into a target biomolecule is through metabolic labeling. Cells are immersed in a medium where access to nutrients is limited to synthetically modified analogues of standard fuels such as sugars. As a consequence, these altered biomolecules are incorporated into the cells in the same manner as their wild-type brethren. The optical probe is then incorporated into the system to image the fate of the altered biomolecules. Other methods of functionalization include enzymatically inserting azides into proteins,[71] and synthesizing phospholipids conjugated to cyclooctynes.[72]
Future directions
As these bioorthogonal reactions are further optimized, they will likely be used for increasingly complex interactions involving multiple different classes of biomolecules. More complex interactions have a smaller margin for error, so increased reaction efficiency is paramount to continued success in optically probing cellular machinery. Also, by minimizing side reactions, the experimental design of a minimally perturbed living system is closer to being realized.
Discovery of biomolecules through metagenomics
The advances in modern sequencing technologies in the late 1990s allowed scientists to investigate DNA of communities of organisms in their natural environments, so-called "eDNA", without culturing individual species in the lab. This metagenomic approach enabled scientists to study a wide selection of organisms that were previously not characterized due in part to an incompetent growth condition. These sources of eDNA include, but are not limited to, soils, ocean, subsurface, hot springs, hydrothermal vents, polar ice caps, hypersaline habitats, and extreme pH environments.[73] Of the many applications of metagenomics, chemical biologists and microbiologists such as Jo Handelsman, Jon Clardy, and Robert M. Goodman who are pioneers of metagenomics, explored metagenomic approaches toward the discovery of biologically active molecules such as antibiotics.[74]

Functional or homology screening strategies have been used to identify genes that produce small bioactive molecules. Functional metagenomic studies are designed to search for specific phenotypes that are associated with molecules with specific characteristics. Homology metagenomic studies, on the other hand, are designed to examine genes to identify conserved sequences that are previously associated with the expression of biologically active molecules.[75]
Functional metagenomic studies enable scientists to discover novel genes that encode biologically active molecules. These assays include top agar overlay assays where antibiotics generate zones of growth inhibition against test microbes, and pH assays that can screen for pH change due to newly synthesized molecules using pH indicator on an agar plate.[76] Substrate-induced gene expression screening (SIGEX), a method to screen for the expression of genes that are induced by chemical compounds, has also been used to search genes with specific functions.[76] These led to the discovery and isolation of several novel proteins and small molecules. For example, the Schipper group identified three eDNA derived AHL lactonases that inhibit biofilm formation of Pseudomonas aeruginosa via functional metagenomic assays.[77] However, these functional screening methods require a good design of probes that detect molecules being synthesized and depend on the ability to express metagenomes in a host organism system.[76]
In contrast, homology metagenomic studies led to a faster discovery of genes that have homologous sequences as the previously known genes that are responsible for the biosynthesis of biologically active molecules. As soon as the genes are sequenced, scientists can compare thousands of bacterial genomes simultaneously.[75] The advantage over functional metagenomic assays is that homology metagenomic studies do not require a host organism system to express the metagenomes, thus this method can potentially save the time spent on analyzing nonfunctional genomes. These also led to the discovery of several novel proteins and small molecules. For example, Banik et al. screened for clones containing genes associated with the synthesis of teicoplanin and vancomycin-like glycopeptide antibiotics and found two new biosynthetic gene clusters.[78] In addition, an in silico examination from the Global Ocean Metagenomic Survey found 20 new lantibiotic cyclases.[79]
There are challenges to metagenomic approaches to discover new biologically active molecules. Only 40% of enzymatic activities present in a sample can be expressed in E. coli..[80] In addition, the purification and isolation of eDNA is essential but difficult when the sources of obtained samples are poorly understood. However, collaborative efforts from individuals from diverse fields including bacterial genetics, molecular biology, genomics, bioinformatics, robots, synthetic biology, and chemistry can solve this problem together and potentially lead to the discovery of many important biologically active molecules.[75]
Protein phosphorylation
Posttranslational modification of proteins with phosphate groups has proven to be a key regulatory step throughout all biological systems. Phosphorylation events, either phosphorylation by protein kinases or dephosphorylation by phosphatases, result in protein activation or deactivation. These events have an immense impact on the regulation of physiological pathways, which makes the ability to dissect and study these pathways integral to understanding the details of cellular processes. There exist a number of challenges—namely the sheer size of the phosphoproteome, the fleeting nature of phosphorylation events and related physical limitations of classical biological and biochemical techniques—that have limited the advancement of knowledge in this area. A recent review[81] provides a detailed examination of the impact of newly developed chemical approaches to dissecting and studying biological systems both in vitro and in vivo.
Through the use of a number of classes of small molecule modulators of protein kinases, chemical biologists have been able to gain a better understanding of the effects of protein phosphorylation. For example, nonselective and selective kinase inhibitors, such as a class of pyridinylimidazole compounds described by Wilson, et al.,[82] are potent inhibitors useful in the dissection of MAP kinase signaling pathways. These pyridinylimidazole compounds function by targeting the ATP binding pocket. Although this approach, as well as related approaches,[83][84] with slight modifications, has proven effective in a number of cases, these compounds lack adequate specificity for more general applications. Another class of compounds, mechanism-based inhibitors, combines detailed knowledge of the chemical mechanism of kinase action with previously utilized inhibition motifs. For example, Parang, et al. describe the development of a "bisubstrate analog" that inhibits kinase action by binding both the conserved ATP binding pocket and a protein/peptide recognition site on the specific kinase.[85] While there is no published in vivo data on compounds of this type, the structural data acquired from in vitro studies have expanded the current understanding of how a number of important kinases recognize target substrates. Interestingly, many research groups utilized ATP analogs as a chemical probe to study kinases and identify their substrates.[86][87][88]
The development of novel chemical means of incorporating phosphomimetics into proteins has provided important insight into the effects of phosphorylation events. Historically, phosphorylation events have been studied by mutating an identified phosphorylation site (serine, threonine or tyrosine) to an amino acid, such as alanine, that cannot be phosphorylated. While this approach has been successful in some cases, mutations are permanent in vivo and can have potentially detrimental effects on protein folding and stability. Thus, chemical biologists have developed new ways of investigating protein phosphorylation. By installing phospho-serine, phospho-threonine or analogous phosphonate mimics into native proteins, researchers are able to perform in vivo studies to investigate the effects of phosphorylation by extending the amount of time a phosphorylation event occurs while minimizing the often-unfavorable effects of mutations. Protein semisynthesis, or more specifically expressed protein ligation (EPL), has proven to be successful techniques for synthetically producing proteins that contain phosphomimetic molecules at either the C- or the N-terminus.[41] In addition, researchers have built upon an established technique in which one can insert an unnatural amino acid into a peptide sequence by charging synthetic tRNA that recognizes a nonsense codon with an unnatural amino acid.[89] Recent developments indicate that this technique can also be employed in vivo, although, due to permeability issues, these in vivo experiments using phosphomimetic molecules have not yet been possible.[90]
Advances in chemical biology have also improved upon classical techniques of imaging kinase action. For example, the development of peptide biosensors—peptides containing incorporated fluorophore molecules—allowed for improved temporal resolution in in vitro binding assays.[91] Experimental limitations, however, prevent this technique from being effectively used in vivo. One of the most useful techniques to study kinase action is Fluorescence Resonance Energy Transfer (FRET). To utilize FRET for phosphorylation studies, fluorescent proteins are coupled to both a phosphoamino acid binding domain and a peptide that can by phosphorylated. Upon phosphorylation or dephosphorylation of a substrate peptide, a conformational change occurs that results in a change in fluorescence.[92] FRET has also been used in tandem with Fluorescence Lifetime Imaging Microscopy (FLIM)[93] or fluorescently conjugated antibodies and flow cytometry[94] to provide a detailed, specific, quantitative results with excellent temporal and spatial resolution.
Through the augmentation of classical biochemical methods as well as the development of new tools and techniques, chemical biologists have improved accuracy and precision in the study of protein phosphorylation.
Metal complexes in medicine
Metal complexes have many characteristics that can be advantageous in drug design. In comparison to organic-based medicines, metal complexes have many more Coordination Numbers, geometries, and oxidation/reduction states that can be used to make structures that interact with targets in unique ways unavailable to most organic molecules. In addition, the cationic metal is advantageous in complexing with charged targets within biological systems like the phosphate backbone of DNA. Targets of metal-based medicines include DNA, proteins, and enzymes. Each target tupe is described in turn below.
Metal complexes targeting DNA
DNA has been the primary target of metal complexes due to the ability of cationic metal interacting with the anionic backbone of DNA. The anticancer chemotherapy drug cisplatin covalently binds to DNA, which disrupts transcription and leads to programmed cell death.[95] Assuming early detection, cisplatin cures almost all cases of testicular cancer. This drug, however, has severe side effects and great effort is being made to improve drug delivery including attachment to single-walled carbon nanotubes,[96] encapsulation in proteins cages,[97] among other clever strategies.
Another major effort for anticancer metal-based drugs centers around stabilization of the G-quadruplex of DNA. In general, these drugs have a non-covalent interaction with the G-quadruplex as well as a planar positively charged structure.[95]
Metal complexes targeting enzymes and proteins
Though DNA has been a primary target for inorganic medicines, enzymes, and proteins also can be modulated through interactions with these compounds. Metal complexes can interact with the amino acids with the highest reduction potential (histidine, cysteine, and selenocysteine).[95] Metals used in such complexes include gold, platinum, ruthenium, vanadium, cobalt and others. Several new potential therapeutic complexes are currently in the process of discovery and investigation.
Gold
Some gold complexes are showing potential as medicines. A rheumatoid arthritis drug (auranofin, a gold(I) phosphine complex) has shown value in treating parasitic disease through inhibiting thioredoxin glutathione reductase.[98]
Platinum
Along with cisplatin, many other platinum complexes are potential therapeutics. Like auranofin, terpyridine platinum inhibits thioredoxin reductase[99] with nanomolar IC50.[100] This complex also is an inhibitor of the common target enzyme topoisomerase I.[99] Yet another family of complexes with potential anticancer properties are dichloro(SMP)-platinum(II) complexes. These complexes target the matrix metalloproteinase, where the complex coordinates with amino acids of the enzyme in the coordination sites previously held by chlorides, and through the smp ligand.[101] As seen by these few examples, platinum complexes are a particularly active area of research for metal-based medicines.
Ruthenium
Ruthenium complexes have anticancer activity. A library of glutathione transferase inhibitors were created through a combination of ethacrynic acid (a known inhibitor of the enzyme) and ruthenium complexes.[102]
Vanadium
Vanadium complexes have been used in multiple therapeutic settings. A new area in which vanadium may have a great medicinal impact is through the oxovanadium porphyrin complexes. These complexes have demonstrated HIV-1 reverse transcriptase inhibition in vitro.[103]
Issues and outlook
Though there is currently much excitement in the field of metal-based medicines, many challenges still face researchers. One such challenge is selectivity of complexes in vivo. Many of these complexes can bind to common proteins like serum albumin in addition to other proteins with amino acids that are common in protein–metal complex interactions like histidine, cysteine, and selenocysteine.[95] Along with selectivity issues, much is yet unknown about mechanisms through which metal complexes interact with proteins. How complexing between a given metal complex and target protein or enzyme occurs is often unknown or unclear and requires much more elucidation before truly effective metal complexes can be designed and delivered.[95] Currently, physicians utilize very few metal-based medicines in the clinics. For example, none of the 21 drugs approved by the U.S. Food and Drug Administration (FDA) in 2008 were inorganic.[104] However, with the success of cisplatin in cancer treatment, it is not unreasonable to anticipate more metal complexes will be actively used in the treatment of diseases.
Synthetic biology
Synthetic biology focuses on the manipulation of biological components to form new systems or the generation of living systems with synthetic parts. The canonical idea of synthetic biology is the creation of new life, but recently it has come to include bioengineering in terms of the use of interchangeable components to give novel outputs. In the search for modular parts, it is most facile if the building blocks contribute independently to the function of the whole unit so that the modules can be recombined in predictable ways. It is useful for synthetic biologists to define "life": in this context, to be alive an organism must be capable of Darwinian evolution – genetic mutation, self-replication and inheritance of mutations.[105]
Synthetic cells
J. Craig Venter's group has created the first "synthetic" cell – the first cells to exist with fully synthetic DNA. Venter was able to manipulate the synthetic genome to dictate the proteins expressed in the organism. Note that these were not fully synthetic cells but that the synthetic DNA was able to take over all metabolic processes necessary for cell survival and proliferation.[106]
DNA as interchangeable parts
DNA is composed of repeating modular units consisting of an anion phosphate group that forms the polyanion backbone, and nucleotide base pairs that engage in Watson-Crick base pairing to form the double strand. Because the molecular recognition of DNA is based mostly on the polyanion backbone, the nucleotides can be modified without altering the structural integrity of the DNA. Steven Benner's group has generated an artificial genetic alphabet of eight new base pairs that can be amplified by polymerase chain reaction; this indicates that these base pairs can be used in systems that undergo Darwinian evolution.[107]
Proteins as interchangeable parts
Amino acids
Amino acids are poor modular building-blocks because they do not act independently and there is a fundamental lack of understanding about the relationship between linear amino acid sequences and the folding and functionality of proteins. Chemical biologists have been able to model, design, and synthesize peptides and evaluate their function.[108]
Protein secondary structure
Modules consisting of protein secondary structure can be designed to perform specific functions; for example, it has been demonstrated that alpha helices can be used as functional peptide catalysts. The Ghadiri group has created a template peptide that promotes the ligation of two modified helices by bringing the helices into close proximity by specifically designed hydrophobic interactions of the helices with the template.[108][109]
Folded proteins
Fully folded proteins can be combined in novel ways to generate specific non-natural outcomes. This is highly useful commercially from drug development to the production of polymers – one can imagine the economic benefits if scientists can design systems in which proteins catalyze reactions without the necessity of excessive human intervention to produce commercially relevant materials. For example, the Keasling group has developed a series of proteins that catalyze conversion of acetyl CoA, a common cellular metabolite, into a precursor for the potent antimalarial drug artemisinin.[110]
Modifying molecular switches
Signaling pathways can be modified to be turned on or off by non-natural ligands or inputs to the system. For instance, systems can be modified so that they are autoinhibited by non-natural proteins that release their inhibition upon binding with a specific molecule that is different from the natural signaling molecule of the path.[111][112] This allows new approaches to studying signal circuits specifically and with user-designed inputs.
Chemical approaches to stem-cell biology
Advances in stem-cell biology have typically been driven by discoveries in molecular biology and genetics. These have included optimization of culture conditions for the maintenance and differentiation of pluripotent and multipotent stem-cells and the deciphering of signaling circuits that control stem-cell fate. However, chemical approaches to stem-cell biology have recently received increased attention due to the identification of several small molecules capable of modulating stem-cell fate in vitro.[113] A small molecule approach offers particular advantages over traditional methods in that it allows a high degree of temporal control, since compounds can be added or removed at will, and tandem inhibition/activation of multiple cellular targets.
Small molecules that modulate stem-cell behavior are commonly identified in high-throughput screens. Libraries of compounds are screened for the induction of a desired phenotypic change in cultured stem-cells. This is usually observed through activation or repression of a fluorescent reporter or by detection of specific cell surface markers by FACS or immunohistochemistry. Hits are then structurally optimized for activity by the synthesis and screening of secondary libraries. The cellular targets of the small molecule can then be identified by affinity chromatography, mass spectrometry, or DNA microarray.
A trademark of pluripotent stem-cells, such as embryonic stem-cells (ESCs), is the ability to self-renew indefinitely. The conventional use of feeder cells and various exogenous growth factors in the culture of ESCs presents a problem in that the resulting highly variable culture conditions make the long-term expansion of un-differentiated ESCs challenging.[114] Ideally, chemically defined culture conditions could be developed to maintain ESCs in a pluripotent state indefinitely. Toward this goal, the Schultz and Ding labs at the Scripps Research Institute identified a small molecule that can preserve the long-term self-renewal of ESCs in the absence of feeder cells and other exogenous growth factors.[115] This novel molecule, called pluripotin, was found to simultaneously inhibit multiple differentiation inducing pathways.
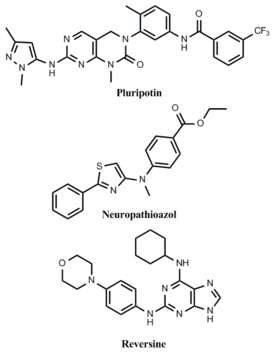
The utility of stem-cells is in their ability to differentiate into all cell types that make up an organism. Differentiation can be achieved in vitro by favoring development toward a particular cell type through the addition of lineage specific growth factors, but this process is typically non-specific and generates low yields of the desired phenotype. Alternatively, inducing differentiation by small molecules is advantageous in that it allows for the development of completely chemically defined conditions for the generation of one specific cell type. A small molecule, neuropathiazol, has been identified which can specifically direct differentiation of multipotent neural stem cells into neurons.[116] Neuropathiazol is so potent that neurons develop even in conditions that normally favor the formation of glial cells, a powerful demonstration of controlling differentiation by chemical means.
Because of the ethical issues surrounding ESC research, the generation of pluripotent cells by reprogramming existing somatic cells into a more "stem-like" state is a promising alternative to the use of standard ESCs. By genetic approaches, this has recently been achieved in the creation of ESCs by somatic cell nuclear transfer[117] and the generation of induced pluripotent stem-cells by viral transduction of specific genes.[118] From a therapeutic perspective, reprogramming by chemical means would be safer than genetic methods because induced stem-cells would be free of potentially dangerous transgenes.[119] Several examples of small molecules that can de-differentiate somatic cells have been identified. In one report, lineage-committed myoblasts were treated with a compound, named reversine, and observed to revert to a more stem-like phenotype.[120] These cells were then shown to be capable of differentiating into osteoblasts and adipocytes under appropriate conditions.[121]
Stem-cell therapies are currently the most promising treatment for many degenerative diseases. Chemical approaches to stem-cell biology support the development of cell-based therapies by enhancing stem-cell growth, maintenance, and differentiation in vitro. Small molecules that have been shown to modulate stem-cell fate are potential therapeutic candidates and provide a natural lean-in to pre-clinical drug development. Small molecule drugs could promote endogenous stem-cells to differentiate, replacing previously damaged tissues and thereby enhancing the body's own regenerative ability. Further investigation of molecules that modulate stem-cell behavior will only unveil new therapeutic targets.
Fluorescence for assessing protein location and function
Fluorophores and techniques to tag proteins
Organisms are composed of cells that, in turn, are composed of macromolecules, e.g. proteins, ribosomes, etc. These macromolecules interact with each other, changing their concentration and suffering chemical modifications. The main goal of many biologists is to understand these interactions, using MRI, ESR, electrochemistry, and fluorescence among others. The advantages of fluorescence reside in its high sensitivity, non-invasiveness, safe detection, and ability to modulate the fluorescence signal. Fluorescence was observed mainly from small organic dyes attached to antibodies to the protein of interest. Later, fluorophores could directly recognize organelles, nucleic acids, and important ions in living cells. In the past decade, the discovery of green fluorescent protein (GFP), by Roger Y. Tsien, hybrid system and quantum dots have enable assessing protein location and function more precisely.[122] Three main types of fluorophores are used: small organic dyes, green fluorescent proteins, and quantum dots. Small organic dyes usually are less than 1 kD, and have been modified to increase photostability, enhance brightness, and reduce self-quenching. Quantum dots have very sharp wavelength, high molar absorptivity and quantum yield. Both organic dyes and quantum dyes do not have the ability to recognize the protein of interest without the aid of antibodies, hence they must use immunolabeling. Since the size of the fluorophore-targeting complex typically exceeds 200 kD, it might interfere with multiprotein recognition in protein complexes, and other methods should be use in parallel. An advantage includes diversity of properties and a limitation is the ability of targeting in live cells. Green fluorescent proteins are genetically encoded and can be covalently fused to your protein of interest. A more developed genetic tagging technique is the tetracysteine biarsenical system, which requires modification of the targeted sequence that includes four cysteines, which binds membrane-permeable biarsenical molecules, the green and the red dyes "FlAsH" and "ReAsH", with picomolar affinity. Both fluorescent proteins and biarsenical tetracysteine can be expressed in live cells, but present major limitations in ectopic expression and might cause lose of function. Giepmans shows parallel applications of targeting methods and fluorophores using GFP and tetracysteine with ReAsH for α-tubulin and β-actin, respectively. After fixation, cells were immunolabeled for the Golgi matrix with QD and for the mitochondrial enzyme cytochrome with Cy5.[122]
Protein dynamics
Fluorescent techniques have been used assess a number of protein dynamics including protein tracking, conformational changes, protein–protein interactions, protein synthesis and turnover, and enzyme activity, among others.
Three general approaches for measuring protein net redistribution and diffusion are single-particle tracking, correlation spectroscopy and photomarking methods. In single-particle tracking, the individual molecule must be both bright and sparse enough to be tracked from one video to the other. Correlation spectroscopy analyzes the intensity fluctuations resulting from migration of fluorescent objects into and out of a small volume at the focus of a laser. In photomarking, a fluorescent protein can be dequenched in a subcellular area with the use of intense local illumination and the fate of the marked molecule can be imaged directly. Michalet and coworkers used quantum dots for single-particle tracking using biotin-quantum dots in HeLa cells.[123]
One of the best ways to detect conformational changes in proteins is to sandwich said protein between two fluorophores. FRET will respond to internal conformational changes result from reorientation of the fluorophore with respect to the other. Dumbrepatil sandwiched an estrogen receptor between a CFP (cyan fluorescent protein) and a YFP (yellow fluorescent protein) to study conformational changes of the receptor upon binding of a ligand.[124]
Fluorophores of different colors can be applied to detect their respective antigens within the cell. If antigens are located close enough to each other, they will appear colocalized and this phenomenon is known as colocalization.[125] Specialized computer software, such as CoLocalizer Pro, can be used to confirm and characterize the degree of colocalization.
FRET can detect dynamic protein–protein interaction in live cells providing the fluorophores get close enough. Galperin et al. used three fluorescent proteins to study multiprotein interactions in live cells.[126]
Tetracysteine biarsenical systems can be used to study protein synthesis and turnover, which requires discrimination of old copies from new copies. In principle, a tetracysteine-tagged protein is labeled with FlAsH for a short time, leaving green labeled proteins. The protein synthesis is then carried out in the presence of ReAsH, labeling the new proteins as red.[127]
One can also use fluorescence to see endogenous enzyme activity, typically by using a quenched activity based proteomics (qABP). Covalent binding of a qABP to the active site of the targeted enzyme will provide direct evidence concerning if the enzyme is responsible for the signal upon release of the quencher and regain of fluorescence.[128]
The unique combination of high spatial and temporal resolution, nondestructive compatibility with living cells and organisms, and molecular specificity insure that fluorescence techniques will remain central in the analysis of protein networks and systems biology.[122]
Applications of DNA microarrays in chemical biology
Planar surfaces functionalized with single- or double-strand nucleic acids have enabled researchers to address a variety of salient biological and biochemical questions in recent years. The general architecture of modern DNA microarrays reflects the historical progression from the sequence-specific probing of whole chromosomes immobilized on glass slides (as early as 1961 with fluorescent in situ hybridization) and the low-density porous membrane arrays available since the early 1990s, to the high-density (102-104 features/mm2) solid support platforms that exist today.[129][130] The massively parallel processing capabilities of these picomolar-range contemporary arrays provide for the generation of large data sets and multiplexed analysis. Furthermore, several top-down and bottom-up assembly methodologies provide researchers with the option for "in-house" production of arrays from custom oligonucleotide libraries[131] or the use of commercial genome chips, notably those developed by Affymetrix and Agilent Technologies.
DNA microarrays can be used to conduct several general types of experiments, most of which relying on the hybridization of fluorescently labeled single-strand DNA molecules isolated from a biological sample to their single-strand complement probes presented on an array. One of the earliest conceived applications for DNA microarrays was for single-nucleotide polymorphism (SNP) genotyping. Since SNPs are a "quick and dirty" approach to detect genetic indicators of pathologies and lineages, arrays in theory provide a facile method for diagnosis; this was confirmed experimentally in the late 1990s in the successful SNP analysis of human tumors.[132] Although there are currently commercially available arrays (e.g. bovine mapping chips) to characterize SNPs, it seems likely that the nascent availability of high-throughput and low-cost pyrosequencing will become the preferred method of recognition, or replace the need for SNP detection altogether with rapid whole-genome sequencing.[133]
A different application of microarray technology that has become the gold standard for RNA analysis in recent years is the widespread utilization of expression microarrays, or "gene chips".[129][130] Gene chip preparation calls for the quantitative reverse transcription of the total cellular RNA pool into labeled and fragmented single-strand DNA prior to hybridization-based capture. Up- and down-regulation of genes in response to stressors or disease states are quantitatively compared in cell lines and organisms.[134] Coupled expression microarray and quantitative proteomics experiments have allowed for the in-depth exploration of the oftentimes non-linear relationship between the abundance of a particular transcribed message and that of its corresponding translated protein.[135] These integrative studies, partially enabled by quantitative DNA microarray technology, have been successfully applied to a variety of biological systems, including yeast,[135] bovine,[136] mouse,[137] bacterial,[138] and human.[139] The expression analysis community has amassed such a significant amount of expression microarray data that they are freely available in public databases.
These types of surfaces can also be used to analyze DNA-protein interactions on a genome-wide scale via chromatin immunoprecipitation, followed by an array-based analysis of the DNA (ChIP-chip). ChIP-chip experiments are enabled by the co-purification of a DNA-binding protein of interest with its corresponding genomic loci when a cross-linked chromatin extract is probed with an antibody to said protein. After purification, amplification and labeling, the DNA is applied to a microarray representing the entire genome; the data are plotted as a histogram that resolves the specific genomic regions associated with that protein. ChIP-chip experiments have provided the scientific community with a wealth of information about the steady-state genomic locations of DNA-binding proteins, such as histones,[140] transcription factors,[141] and polymerase machinery,[142] and have also been successfully applied to studies on the dynamics of transcription factor binding.[143] The data from these experiments may be further manipulated to computationally derive consensus binding sequences for some transcription factors, giving the opportunity for insight into the in vivo behavior of the factor, deeper than simple information about localization.[144]
DNA microarrays are also amenable to the direct analysis of protein–DNA interactions in kinetic binding assays as analyzed by surface plasmon resonance (SPR).[145] This experimental approach also relies on single-strand DNA immobilized on a high-density array; however, the quantitative readout is based on a change in the optical properties of the DNA-functionalized surface when a protein flowed over the surface binds to the sequence in a particular surface feature. DNA-functionalized arrays analyzed with SPR in this way have yielded kinetic data regarding fundamental molecular biological processes. Recently, SPR analysis of a DNA microarray and components of the DNA replication machinery helped to elucidate the biochemical nuances of the replication fork.[146]
High-density DNA microarrays have emerged as an important component of the chemical biology toolkit. The existing technology allows for the construction of customizable, as well as general, arrays and provides researchers with the opportunity to generate robust data from many different types of biological inputs. Considering the relatively recent shift in the scientific community away from binary perturbation/readout studies and toward "big science" and large data sets, it seems likely that DNA microarrays will continue to enable pertinent biological research for many years to come.
Applications of Chemical Biology in Drug Discovery
Chemical biology approaches can help answer important questions of relevance to small molecule drug discovery projects. This includes questions related to the characterization of protein targets and the molecular pharmacology of small molecule drugs that modulate target function.[147]
Chemical Biology Approaches to Characterize Protein Targets
| | ||
|
Question |
Why ask? |
Chemical Biology Technique |
|
What is my target? |
Enables target-based drug discovery on hits/mechanisms from phenotypic screens. |
Affinity chemoproteomics; Mutagenesis; Phenotypic screening; Chemogenomics; Proteomics |
|
What is the subcellular distribution of my target? |
Location of target may influence screening assays or inhibitor design; active and inactive species of the target may localize to different cellular regions; target may be activated by particular environment owing to localization to a particular organelle (for example, acidic lysosome) |
Imaging; Microscopy |
|
Does my target exist in multiple forms, and does this vary across tissues and species? |
May reveal species differences, splice variants, relevance of full length target vs. catalytic domains for screening; splice variants of the target may have different protein domains, activity, cellular location, tissue distribution, and affinity for substrate; knowing correct sequence cDNA enables potential to express recombinant protein or to generate overexpression cell line which can inform choice of primary assay and screening sequence. |
Computational biology; Genotype-tissue-expression analysis; Proteomics |
|
What is the endogenous ligand for my target and its concentration in the diseased state? |
Allows one to understand what biological pathways are being modulated (one of the keys for establishing a physiologically relevant assay); allows one to theorize what will happen if endogenous substrate levels are increased by inhibiting the desired target. |
Immunoprecipitation; Metabolomics; Peptide Microarrays; Peptidomics |
|
How well characterized is the interaction of the endogenous substrate with my target? |
Informs chemical feasibility, screening strategy and medicinal chemistry approach (for example, substrate concentration and Km for enzyme target) |
Biochemical enzyme and cellular activity assays |
|
Is my target post-translationally modified? |
Can rationalize differences in affinity/efficacy between biochemical in vitro and cellular/in vivo systems; informs primary assay choice and screen sequence. |
Chemical Probe; Immunodetection; Metabolomics; Proteomics; Phosphoproteomics |
|
What other proteins are influential in regulating substrate concentrations? |
Can represent alternate strategies for modulating the desired pathway. Can lead to complimentary pharmacology. |
Computational Biology; RNAi |
|
What is the turnover of my target and is this affected by my compound? |
Especially important to understand the cellular efficiencies of covalent modalities. |
Chemical probe; Immunoprecipitation; Pulse/chase; SILAC/MS |
|
What is the abundance of my target, and does it vary? |
Abundance of a protein might vary by tissue, disease state, or as a response to drug action. |
Targeted quantitative proteomics coupled with immunocapture |
|
Does my target interact with other proteins and what is the consequence of these interactions? |
Can lead to a better understanding of signaling pathway; informs screening assay; could be important that protein forms hetero or homodimers. |
Immunoprecipitation |
|
What potential off-targets are most closely related to my target sequence and function? |
Informs screening sequence design; important to consider not only targets that are closely related in terms of binding site sequence, but also those most closely related in a chemogenomic sense; understand which tissues express off-targets. |
Computational Biology |
Protein Target Characterization
- What is my target?
- - Affinity chemoproteomics can be used to identify the targets of compounds that have shown activity in a phenotypic or pathway screen. For example, this approach was used to identify of MTH1 as a potential anticancer target of the (S) form of Crizotinib.[148] Affinity chemoproteomics technology was also used to identify the BET bromodomains as the molecular targets of a series of small molecule modulators of Apolipoprotein A1 (ApoA1).[149]
- - Mutagensis is important target validation approach is to generate a catalytically-dead target protein to tease apart the role of the enzymatic and scaffolding functions. This method is often used in kinase validation experiments where the pharmacological performance of a putative inhibitor is tested in cells with the kinase knocked out and replaced through transfection of the recombinant wild-type protein or its kinase dead mutant. This approach was used recently to confirm DCLK1 as the functional anti-proliferative target of a previously developed LRRK2 inhibitor for Parkinson's disease[150]
- Does my target exist in multiple related forms and does this vary across different tissues and species?
- - Genotype-tissue expression project (GTEx) has generated a database of mRNA expression levels across multiple tissues. An illustration of the use of this resource shows that PDE4B is expressed in multiple forms that vary across human tissues. Being aware of these different forms has enabled development of a selective inhibitor targeting PDE4B in the brain through taking advantage of differences in sequences in the long and short forms outside the active site.[151]
- What is the endogenous ligand for my target and its concentration in the diseased state?
- - Metabolomics in combination with clickable chemical reporters can enable chemoproteomic methods to be applied to substrate identification, as shown recently for protein lipidation.[152]
- Is my target post-translationally modified?
- - Dimedone chemical probes that chemoselectively react with sulfenic acid residues in EGFR have shown how cysteine oxidation can be use for enzymatic regulation.[153]
- What is the turnover of my target and is this affected by my compound?
- - Chemical probes of BTK were used to confirm its slow turnover rate.[154]
- - Immunoprecipitation of EGFR has been used to confirm increased protein half-livey, likely due to a concomitant decrease in binding to the Cbl ubiquitin ligase responsible for regulating the degradation of EGFR.[155] This increased half-life could result in a need to change the projected dose or frequency. In addition to mutations altering the turnover rate of a protein, small molecule drugs can also change the turnover and thus the amount of protein in the cell. For instance, many kinases are known to be clients of the Hsp90-Cdc37 chaperone system and recent studies have found that some ATP-competitive inhibitors disrupt the ability of Cdc37 to bind to the target kinase and recruit it to Hsp9015.[156]
- What potential off- targets are most closely related to my target sequence and function?
- - Computational biology and structural bioinformatic analyses of protein sequences and binding pocket topology can be used to delineate which proteins are similar to a desired target's biological sequence as well as its binding site shape. Additionally, chemoinformatic approaches using in silico tools to predict potential off-targets based on the structural similarity between a novel small molecule and previously known compounds with different pharmacology are also useful.
Chemical Biology Approaches to Characterize Molecular Pharmacology
| | ||
|
Question |
Why ask? |
Chemical Biology Technique |
|
What target(s) and off-targets does my molecule bind? |
Key to understanding what drives molecule efficacy and potential safety liabilities; allows team to understand mechanism of action, pathways affected and target that drives efficacy; enables target-based drug discovery and understanding or improvement of potency and selectivity. |
Activity probes; Affinity capture; ABPP; DARTS; Photoaffinity; Proteomics; Protein microarrays; SPROX, Thermal aggregation; Y3H assays |
|
Where does my molecule bind? |
Characterization of binding kinetics and binding site may be key to functional translation and pharmacokinetic-pharmacodynamic relationships. |
Hydrogen-deuterium exchange |
|
How does my molecule bind? |
Characterization of binding kinetics and binding site may be key to functional translation and PK/PD. |
Kinetics |
|
What is the tissue distribution of my molecule? |
Assuming plasma exposure reflects target tissue exposure is often incorrect. Affects Pillar 1. |
MALDI MS; PK: Protein binding; Tissue distribution |
|
What are the functional consequences to my target when my molecule binds? |
Provides an understanding of how the molecule works; binding could modulate target degradation, stabilization, translocation or interactions with other proteins. |
Immunodetection; Proteomics |
|
How much target occupancy do I need to drive my relevant biological phenotype? |
Allows team to develop Pillar II confidence and link to Pillar III. Key enabler to help define Ceff. |
Occupancy probes; "Three pillars in a tube" |
Molecular Pharmacology Characterization
- What target(s) and off-targets does my molecule bind?
- - Activity probes such as nucleotide acyl phosphates (KiNativ™) probes that react chemoselectively with the catalytic lysine of >80% of all known kinases can be used to determine many of the kinase targets of a molecule.[157]
- - ABPP using probes derived from covalent kinase inhibitors, have been used to determine the cellular targets of these covalent inhibitors in living cells.[158]
- - Chemoproteomics was used to assess the multiple binding partners of HDAC inhibitors in protein complexes scaffolded by ELM-SANT domain subunits.[159] Thermal proteomics, where one looks for proteins that show increased thermal stability at elevated temperatures due to compound binding, has been used to identify off-targets of several kinase inhibitors including the BRAF inhibitor Vemurafenib.[160] Affinity capture where a chemical probe is tethered to a solid support, was used to identify cereblon (CRBN) as a thalidomide binding protein.[161]
- - DARTS (Drug Affinity Responsive Target Stability); takes advantage of a reduction in the protease susceptibility of the target protein upon drug binding. This technique was used to confirm binding biological binding partners for a number of molecules: rapamycin and FKBP with FKBP12 and resveratrol with eIF4A.[162]
- - SPROX (Stability of Proteins from Rates of Oxidation); utilizes the chemical denaturant-dependent oxidation rates of methionine residues in a protein to report on the thermodynamic properties of its global and/or subglobal unfolding/refolding reactions.[163]
- What is the tissue distribution of my molecule?
- - Quantitative mass spectrometry (MS) was recently used to determine the concentrations of the chemotherapeutic agent YM155 in various cancer cells.[164]
- What are the functional consequences to my target when my molecule binds?
- - The use of shRNA gene silencing combined with immunoblotting showed that BRAF inhibitors GDC-0879 and PLX4720 block MAPK signaling in BRAF (V600E) tumors while these same compounds activate the RAF-MEK-ERK pathway in KRAS mutant and RAS/RAF wild-type tumors.[165]
- How much target occupancy do I need to drive my relevant biological phenotype?
- - A clickable covalent occupancy probe of fatty acid amide hydrolase (FAAH) has been used to relate target engagement to anandamide elevation and efficacy in models of rodent inflammatory and neuropathic pain.[166] Target occupancy quantification can also be used to validate the relevance of a drug target identified from phenotypic screening. A clickable covalent probe of the mRNA decapping enzyme DcpS, which used a sulfonyl fluoride warhead to target a reactive tyrosine in the binding site of the enzyme, enabled the assessment of DcpS target engagement of a diaminoquinazoline inhibitor (developed from a phenotypic screen for the treatment of spinal muscular atrophy).[167]
See also
References
- ↑ Cox Jü, Mann M (2007). "Is Proteomics the New Genomics?". Cell. 130 (3): 395–8. doi:10.1016/j.cell.2007.07.032. PMID 17693247.
- ↑ Zhao Y, Jensen ON (October 2009). "Modification-specific proteomics: strategies for characterization of post-translational modifications using enrichment techniques". Proteomics. 9 (20): 4632–41. doi:10.1002/pmic.200900398. PMC 2892724
 . PMID 19743430.
. PMID 19743430. - ↑ Gingras A-C, Gstaiger M, Raught B, Aebersold R (2007). "Analysis of protein complexes using mass spectrometry". Nature Reviews Molecular Cell Biology. 8 (8): 645–54. doi:10.1038/nrm2208. PMID 17593931.
- ↑ Chin JW, Schultz PG (November 2002). "In vivo photocrosslinking with unnatural amino Acid mutagenesis". Chembiochem. 3 (11): 1135–7. doi:10.1002/1439-7633(20021104)3:11<1135::AID-CBIC1135>3.0.CO;2-M. PMID 12404640.
- ↑ Liu W, Brock A, Chen S, Chen S, Schultz PG (2007). "Genetic incorporation of unnatural amino acids into proteins in mammalian cells". Nature Methods. 4 (3): 239–44. doi:10.1038/nmeth1016. PMID 17322890.
- ↑ López-Otín C, Overall CM (2002). "Protease degradomics: A new challenge for proteomics". Nature Reviews Molecular Cell Biology. 3 (7): 509–19. doi:10.1038/nrm858. PMID 12094217.
- ↑ Adam GC, Cravatt BF, Sorensen EJ (January 2001). "Profiling the specific reactivity of the proteome with non-directed activity-based probes". Chem. Biol. 8 (1): 81–95. doi:10.1016/S1074-5521(00)90060-7. PMID 11182321.
- ↑ Tureček F (2002). "Mass spectrometry in coupling with affinity capture-release and isotope-coded affinity tags for quantitative protein analysis". Journal of Mass Spectrometry. 37 (1): 1–14. doi:10.1002/jms.275. PMID 11813306.
- ↑ Blethrow J, Zhang C, Shokat KM, Weiss EL (May 2004). "Design and use of analog-sensitive protein kinases". Curr Protoc Mol Biol. Chapter 18: Unit 18.11. doi:10.1002/0471142727.mb1811s66. PMID 18265343.
- ↑ Bright Ideas for Chemical Biology – ACS Chemical Biology (ACS Publications)
- ↑ The Nobel Prize in Chemistry 2008
- ↑ Uil TG, Haisma HJ, Rots MG (November 2003). "Therapeutic modulation of endogenous gene function by agents with designed DNA-sequence specificities". Nucleic Acids Res. 31 (21): 6064–78. doi:10.1093/nar/gkg815. PMC 275457. PMID 14576293.
- ↑ Kuwabara T, Warashina M, Taira K (July 2002). "Cleavage of an inaccessible site by the maxizyme with two independent binding arms: an alternative approach to the recruitment of RNA helicases". J. Biochem. 132 (1): 149–55. doi:10.1093/oxfordjournals.jbchem.a003193. PMID 12097172.
- ↑ Doudna JA, Cech TR (July 2002). "The chemical repertoire of natural ribozymes". Nature. 418 (6894): 222–8. Bibcode:2002Natur.418..222D. doi:10.1038/418222a. PMID 12110898.
- ↑ Hamilton AJ, Baulcombe DC (October 1999). "A species of small antisense RNA in posttranscriptional gene silencing in plants". Science. 286 (5441): 950–2. doi:10.1126/science.286.5441.950. PMID 10542148.
- ↑ Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (February 1998). "Potent and specific genetic interference by double-strand RNA in Caenorhabditis elegans". Nature. 391 (6669): 806–11. Bibcode:1998Natur.391..806F. doi:10.1038/35888. PMID 9486653.
- ↑ Baulcombe D (June 2005). "RNA silencing". Trends Biochem. Sci. 30 (6): 290–3. doi:10.1016/j.tibs.2005.04.012. PMID 15950871.
- ↑ Xia H, Mao Q, Paulson HL, Davidson BL (October 2002). "siRNA-mediated gene silencing in vitro and in vivo". Nat. Biotechnol. 20 (10): 1006–10. doi:10.1038/nbt739. PMID 12244328.
- ↑ Shim MS, Kwon YJ (December 2010). "Efficient and targeted delivery of siRNA in vivo". FEBS J. 277 (23): 4814–27. doi:10.1111/j.1742-4658.2010.07904.x. PMID 21078116.
- ↑ Dorsett Y, Tuschl T (April 2004). "siRNAs: applications in functional genomics and potential as therapeutics". Nature Reviews Drug Discovery. 3 (4): 318–29. doi:10.1038/nrd1345. PMID 15060527.
- ↑ Mohr S, Bakal C, Perrimon N (2010). "Genomic screening with RNAi: results and challenges". Annu. Rev. Biochem. 79: 37–64. doi:10.1146/annurev-biochem-060408-092949. PMC 3564595. PMID 20367032.
- ↑ Ryther RC, Flynt AS, Phillips JA, Patton JG (January 2005). "siRNA therapeutics: big potential from small RNAs". Gene Ther. 12 (1): 5–11. doi:10.1038/sj.gt.3302356. PMID 15496962.
- ↑ Jordens S, Adamcik J, Amar-Yuli I, Mezzenga R (2011). "Disassembly and Reassembly of Amyloid Fibrils in Water−Ethanol Mixtures". Biomacromolecules. 12 (1): 187–93. doi:10.1021/bm101119t. PMID 21142059.
- ↑ Reddy G, Straub JE, Thirumalai D (December 2010). "Dry amyloid fibril assembly in a yeast prion peptide is mediated by long-lived structures containing water wires". Proc. Natl. Acad. Sci. USA. 107 (50): 21459–64. Bibcode:2010PNAS..10721459R. doi:10.1073/pnas.1008616107. PMC 3003024. PMID 21098298.
- ↑ Borman SA (2010). "Water Factors in on Amyloid And Prion Aggregation Rates". Chemical & Engineering News. 88 (49): 37.
- 1 2 Luheshi LM, Crowther DC, Dobson CM (2008). "Protein misfolding and disease: from the test tube to the organism". Current Opinion in Chemical Biology. 12 (1): 25–31. doi:10.1016/j.cbpa.2008.02.011. PMID 18295611.
- ↑ Luheshi LM, Tartaglia GG, Brorsson A-C, Pawar AP, Watson IE, Chiti F, Vendruscolo M, Lomas DA, Dobson CM (2007). "Systematic in Vivo Analysis of the Intrinsic Determinants of Amyloid β Pathogenicity". PLoS Biology. 5 (11): e290. doi:10.1371/journal.pbio.0050290. PMC 2043051. PMID 17973577.
- ↑ Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FAI, Gubb DC, Lomas DA (2005). "Intraneuronal Aβ, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease". Neuroscience. 132 (1): 123–35. doi:10.1016/j.neuroscience.2004.12.025. PMID 15780472.
- ↑ Hammarström P, Sekijima Y, White JT, Wiseman RL, Lim A, Costello CE, Altland K, Garzuly F, Budka H (2003). "D18G Transthyretin is Monomeric, Aggregation Prone, and Not Detectable in Plasma and Cerebrospinal Fluid: A Prescription for Central Nervous System Amyloidosis?†". Biochemistry. 42 (22): 6656–63. doi:10.1021/bi027319b. PMID 12779320.
- ↑ Sekijima Y, Wiseman RL, Matteson J, Hammarström P, Miller SR, Sawkar AR, Balch WE, Kelly JW (2005). "The Biological and Chemical Basis for Tissue-Selective Amyloid Disease". Cell. 121 (1): 73–85. doi:10.1016/j.cell.2005.01.018. PMID 15820680.
- ↑ Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI (2006). "Progressive Disruption of Cellular Protein Folding in Models of Polyglutamine Diseases". Science. 311 (5766): 1471–1474. Bibcode:2006Sci...311.1471G. doi:10.1126/science.1124514. PMID 16469881.
- ↑ Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A (2006). "Opposing Activities Protect Against Age-Onset Proteotoxicity". Science. 313 (5793): 1604–1610. Bibcode:2006Sci...313.1604C. doi:10.1126/science.1124646. PMID 16902091.
- ↑ Ramirez-Alvarado, M., Kelly, J. W., and Dobson, C. M., (Eds.) (2010) Protein Misfolding Diseases Current and Emerging Principles and Therapies, John Wiley and Sons, Hoboken
- 1 2 Kimmerlin T, Seebach D (2005). "'100 years of peptide synthesis': ligation methods for peptide and protein synthesis with applications to beta-peptide assemblies". J Pept Res. 65 (2): 229–260. doi:10.1111/j.1399-3011.2005.00214.x. PMID 15705167.
- 1 2 Kent S (June 2006). "Obituary: Bruce Merrifield (1921–2006)". Nature. 441 (7095): 824. Bibcode:2006Natur.441..824K. doi:10.1038/441824a. PMID 16778881.
- ↑ Dirksen A, Dawson PE (2008). "Expanding the scope of chemoselective peptide ligations in chemical biology". Current Opinion in Chemical Biology. 12 (6): 760–6. doi:10.1016/j.cbpa.2008.10.009. PMID 19058994.
- ↑ Dawson PE, Muir TW, Clarklewis I, Kent SBH (1994). "SYNTHESIS OF PROTEINS BY NATIVE CHEMICAL LIGATION". Science. 266 (5186): 776–779. Bibcode:1994Sci...266..776D. doi:10.1126/science.7973629. PMID 7973629.
- ↑ Torbeev VY, Kent SB (2007). "Convergent chemical synthesis and crystal structure of a 203 amino acid "covalent dimer" HIV-1 protease enzyme molecule". Angew. Chem. Int. Ed. Engl. 46 (10): 1667–70. doi:10.1002/anie.200604087. PMID 17397076.
- ↑ Durek T, Torbeev VY, Kent SB (March 2007). "Convergent chemical synthesis and high-resolution x-ray structure of human lysozyme". Proc. Natl. Acad. Sci. USA. 104 (12): 4846–51. Bibcode:2007PNAS..104.4846D. doi:10.1073/pnas.0610630104. PMC 1829227. PMID 17360367.
- ↑ McCaldon P, Argos P (1988). "Oligopeptide biases in protein sequences and their use in predicting protein coding regions in nucleotide sequences". Proteins. 4 (2): 99–122. doi:10.1002/prot.340040204. PMID 3227018.
- 1 2 Muir TW, Sondhi D, Cole PA (June 1998). "Expressed protein ligation: a general method for protein engineering". Proc. Natl. Acad. Sci. USA. 95 (12): 6705–10. Bibcode:1998PNAS...95.6705M. doi:10.1073/pnas.95.12.6705. PMC 22605. PMID 9618476.
- ↑ Wu B, Chen J, Warren JD, Chen G, Hua Z, Danishefsky SJ (June 2006). "Building complex glycopeptides: Development of a cysteine-free native chemical ligation protocol". Angew. Chem. Int. Ed. Engl. 45 (25): 4116–25. doi:10.1002/anie.200600538. PMID 16710874.
- ↑ Chatterjee C, McGinty RK, Pellois JP, Muir TW (2007). "Auxiliary-mediated site-specific peptide ubiquitylation". Angew. Chem. Int. Ed. Engl. 46 (16): 2814–8. doi:10.1002/anie.200605155. PMID 17366504.
- ↑ Soellner MB, Nilsson BL, Raines RT (July 2006). "Reaction mechanism and kinetics of the traceless Staudinger ligation". J. Am. Chem. Soc. 128 (27): 8820–8. doi:10.1021/ja060484k. PMID 16819875.
- ↑ Punna S, Kuzelka J, Wang Q, Finn MG (April 2005). "Head-to-tail peptide cyclodimerization by copper-catalyzed azide-alkyne cycloaddition". Angew. Chem. Int. Ed. Engl. 44 (15): 2215–20. doi:10.1002/anie.200461656. PMID 15693048.
- ↑ Dirksen A, Hackeng TM, Dawson PE (November 2006). "Nucleophilic catalysis of oxime ligation". Angew. Chem. Int. Ed. Engl. 45 (45): 7581–4. doi:10.1002/anie.200602877. PMID 17051631.
- ↑ Jäckel C, Kast P, Hilvert D (2008). "Protein design by directed evolution". Annu Rev Biophys. 37: 153–73. doi:10.1146/annurev.biophys.37.032807.125832. PMID 18573077.
- 1 2 Taylor SV, Walter KU, Kast P, Hilvert D (September 2001). "Searching sequence space for protein catalysts". Proc. Natl. Acad. Sci. USA. 98 (19): 10596–601. Bibcode:2001PNAS...9810596T. doi:10.1073/pnas.191159298. PMC 58511. PMID 11535813.
- ↑ Bittker JA, Le BV, Liu JM, Liu DR (May 2004). "Directed evolution of protein enzymes using nonhomologous random recombination". Proc. Natl. Acad. Sci. USA. 101 (18): 7011–6. Bibcode:2004PNAS..101.7011B. doi:10.1073/pnas.0402202101. PMC 406457. PMID 15118093.
- ↑ Aharoni A, Griffiths AD, Tawfik DS (April 2005). "High-throughput screens and selections of enzyme-encoding genes". Curr Opin Chem Biol. 9 (2): 210–6. doi:10.1016/j.cbpa.2005.02.002. PMID 15811807.
- ↑ Wilson DS, Keefe AD, Szostak JW (March 2001). "The use of mRNA display to select high-affinity protein-binding peptides". Proc. Natl. Acad. Sci. USA. 98 (7): 3750–5. Bibcode:2001PNAS...98.3750W. doi:10.1073/pnas.061028198. PMC 31124. PMID 11274392.
- ↑ Tawfik DS, Griffiths AD (1998). "Man-made cell-like compartments for molecular evolution". Nature Biotechnology. 16 (7): 652–6. doi:10.1038/nbt0798-652. PMID 9661199.
- ↑ Lo Surdo P, Walsh MA, Sollazzo M (April 2004). "A novel ADP- and zinc-binding fold from function-directed in vitro evolution". Nature Structural & Molecular Biology. 11 (4): 382–3. doi:10.1038/nsmb745. PMID 15024384.
- ↑ Kamtekar S, Schiffer JM, Xiong HY, Babik JM, Hecht MH (1993). "Protein Design by Binary Patterning of Polar and Nonpolar Amino-acids". Science. 262 (5140): 1680–1685. Bibcode:1993Sci...262.1680K. doi:10.1126/science.8259512. PMID 8259512.
- ↑ Wei Y, Kim S, Fela D, Baum J, Hecht MH (November 2003). "Solution structure of a de novo protein from a designed combinatorial library". Proc. Natl. Acad. Sci. USA. 100 (23): 13270–3. Bibcode:2003PNAS..10013270W. doi:10.1073/pnas.1835644100. PMC 263778. PMID 14593201.
- ↑ Vamvaca K, Butz M, Walter KU, Taylor SV, Hilvert D (2005). "Simultaneous optimization of enzyme activity and quaternary structure by directed evolution". Protein Science. 14 (8): 2103–14. doi:10.1110/ps.051431605. PMC 2279322. PMID 15987889.
- ↑ MacBeath G, Kast P, Hilvert D (1998). "Redesigning enzyme topology by directed evolution". Science. 279 (5358): 1958–1961. Bibcode:1998Sci...279.1958M. doi:10.1126/science.279.5358.1958. PMID 9506949.
- ↑ Yoshikuni Y, Ferrin TE, Keasling JD (2006). "Designed divergent evolution of enzyme function". Nature. 440 (7087): 1078–1082. Bibcode:2006Natur.440.1078Y. doi:10.1038/nature04607. PMID 16495946.
- ↑ Seelig B, Szostak JW (2007). "Selection and evolution of enzymes from a partially randomized non-catalytic scaffold". Nature. 448 (7155): 828–31. Bibcode:2007Natur.448..828S. doi:10.1038/nature06032. PMID 17700701.
- ↑ Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS (1998). "High-resolution protein design with backbone freedom". Science. 282 (5393): 1462–1467. doi:10.1126/science.282.5393.1462. PMID 9822371.
- ↑ Jewett JC, Bertozzi CR (2010). "Cu-free click cycloaddition reactions in chemical biology". Chemical Society Reviews. 39 (4): 1272–9. doi:10.1039/b901970g. PMC 2865253. PMID 20349533..
- ↑ Sletten EM, Bertozzi CR (2009). "Bioorthogonal chemistry: fishing for selectivity in a sea of functionality". Angew. Chem. Int. Ed. Engl. 48 (38): 6974–98. doi:10.1002/anie.200900942. PMC 2864149. PMID 19714693.
- ↑ Kolb HC, Finn MG, Sharpless KB (June 2001). "Click Chemistry: Diverse Chemical Function from a Few Good Reactions". Angew. Chem. Int. Ed. Engl. 40 (11): 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. PMID 11433435.
- ↑ Rostovtsev VV, Green LG, Fokin VV, Sharpless KB (July 2002). "A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes". Angew. Chem. Int. Ed. Engl. 41 (14): 2596–9. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. PMID 12203546.
- ↑ Hong V, Steinmetz NF, Manchester M, Finn MG (2010). "Labeling Live Cells by Copper-Catalyzed Alkyne−Azide Click Chemistry". Bioconjugate Chemistry. 21 (10): 1912–6. doi:10.1021/bc100272z. PMC 3014321. PMID 20886827.
- ↑ Agard NJ, Prescher JA, Bertozzi CR (November 2004). "A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems". J. Am. Chem. Soc. 126 (46): 15046–7. doi:10.1021/ja044996f. PMID 15547999.
- ↑ Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR (October 2007). "Copper-free click chemistry for dynamic in vivo imaging". Proc. Natl. Acad. Sci. USA. 104 (43): 16793–7. Bibcode:2007PNAS..10416793B. doi:10.1073/pnas.0707090104. PMC 2040404. PMID 17942682.
- ↑ Blackman ML, Royzen M, Fox JM (2008). "Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity". Journal of the American Chemical Society. 130 (41): 13518–9. doi:10.1021/ja8053805. PMC 2653060. PMID 18798613.
- ↑ Devaraj NK, Weissleder R, Hilderbrand SA (2008). "Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging". Bioconjugate Chemistry. 19 (12): 2297–9. doi:10.1021/bc8004446. PMC 2677645. PMID 19053305.
- ↑ Pipkorn Rü, Waldeck W, Didinger B, Koch M, Mueller G, Wiessler M, Braun K (2009). "Inverse-electron-demand Diels-Alder reaction as a highly efficient chemoselective ligation procedure: Synthesis and function of a BioShuttle for temozolomide transport into prostate cancer cells". Journal of Peptide Science. 15 (3): 235–41. doi:10.1002/psc.1108. PMID 19177421.
- ↑ Hur GH, Meier JL, Baskin J, Codelli JA, Bertozzi CR, Marahiel MA, Burkart MD (April 2009). "Crosslinking studies of protein–protein interactions in nonribosomal peptide biosynthesis". Chem. Biol. 16 (4): 372–81. doi:10.1016/j.chembiol.2009.02.009. PMC 2743379. PMID 19345117.
- ↑ Neef AB, Schultz C (2009). "Selective fluorescence labeling of lipids in living cells". Angew. Chem. Int. Ed. Engl. 48 (8): 1498–500. doi:10.1002/anie.200805507. PMID 19145623.
- ↑ Keller M, Zengler K (February 2004). "Tapping into microbial diversity". Nature Reviews Microbiology. 2 (2): 141–50. doi:10.1038/nrmicro819. PMID 15040261.
- ↑ Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM (October 1998). "Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products". Chem. Biol. 5 (10): R245–9. doi:10.1016/S1074-5521(98)90108-9. PMID 9818143.
- 1 2 3 Banik JJ, Brady SF (2010). "Recent application of metagenomic approaches toward the discovery of antimicrobials and other bioactive small molecules". Current Opinion in Microbiology. 13 (5): 603–609. doi:10.1016/j.mib.2010.08.012. PMC 3111150. PMID 20884282.
- 1 2 3 Daniel R (2005). "The metagenomics of soil". Nature Reviews Microbiology. 3 (6): 470–478. doi:10.1038/nrmicro1160. PMID 15931165.
- ↑ Schipper C, Hornung C, Bijtenhoorn P, Quitschau M, Grond S, Streit WR (2009). "Metagenome-Derived Clones Encoding Two Novel Lactonase Family Proteins Involved in Biofilm Inhibition in Pseudomonas aeruginosa". Applied and Environmental Microbiology. 75 (1): 224–233. doi:10.1128/AEM.01389-08. PMC 2612230. PMID 18997026.
- ↑ Bunterngsook B, Kanokratana P, Thongaram T, Tanapongpipat S, Uengwetwanit T, Rachdawong S, Vichitsoonthonkul T, Eurwilaichitr L (2010). "Identification and characterization of lipolytic enzymes from a peat-swamp forest soil metagenome". Biosci. Biotechnol. Biochem. 74 (9): 1848–54. doi:10.1271/bbb.100249. PMID 20834152.
- ↑ Li B, Sher D, Kelly L, Shi YX, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW (2010). "Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria". Proceedings of the National Academy of Sciences of the United States of America. 107 (23): 10430–10435. Bibcode:2010PNAS..10710430L. doi:10.1073/pnas.0913677107. PMC 2890784. PMID 20479271.
- ↑ Gabor EM, Alkema WBL, Janssen DB (2004). "Quantifying the accessibility of the metagenome by random expression cloning techniques". Environmental Microbiology. 6 (9): 879–86. doi:10.1111/j.1462-2920.2004.00640.x. PMID 15305913.
- ↑ Tarrant MK, Cole PA (2009). "The Chemical Biology of Protein Phosphorylation". Annual Review of Biochemistry. 78: 797–825. doi:10.1146/annurev.biochem.78.070907.103047. PMC 3074175. PMID 19489734.
- ↑ Wilson KP, McCaffrey PG, Hsiao K, Pazhanisamy S, Galullo V, Bemis GW, Fitzgibbon MJ, Caron PR, Murcko MA, Su MS (June 1997). "The structural basis for the specificity of pyridinylimidazole inhibitors of p38 MAP kinase". Chem. Biol. 4 (6): 423–31. doi:10.1016/S1074-5521(97)90194-0. PMID 9224565.
- ↑ Pargellis C, Ton L, Churchill L, CIrillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N (2002). "Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site". Nature Structural & Molecular Biology. 9 (4): 268–272. doi:10.1038/nsb770. PMID 11896401.
- ↑ Schindler T, Bornmann W, Pellicena P, Miller WT, Clarckson B, Kuriyan J (2000). "Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase". Science. 289 (5486): 1938–1942. Bibcode:2000Sci...289.1938S. doi:10.1126/science.289.5486.1938. PMID 10988075.
- ↑ Parang K, Till JH, Ablooglu AJ, Kohanski RA, Hubbard SR, Cole PA (2001). "Mechanism-based design of a protein kinase inhibitor". Nature Structural & Molecular Biology. 8 (1): 37–41. doi:10.1038/83028. PMID 11135668.
- ↑ Fouda, A. E. and Pflum, M. K. H. (2015), A Cell-Permeable ATP Analogue for Kinase-Catalyzed Biotinylation. Angew. Chem. Int. Ed., 54: 9618–9621. doi:10.1002/anie.201503041
- ↑ Chamara Senevirathne, D. Maheeka Embogama, Thilani A. Anthony, Ahmed E. Fouda, Mary Kay H. Pflum, The generality of kinase-catalyzed biotinylation, Bioorganic & Medicinal Chemistry, Volume 24, Issue 1, 1 January 2016, Pages 12-19, ISSN 0968-0896, http://dx.doi.org/10.1016/j.bmc.2015.11.029. (http://www.sciencedirect.com/science/article/pii/S0968089615301528)
- ↑ Anthony, T. M., Dedigama-Arachchige, P. M., Embogama, D. M., Faner, T. R., Fouda, A. E. and Pflum, M. K. H. (2015) ATP Analogs in Protein Kinase Research, in Kinomics: Approaches and Applications (eds H.-B. Kraatz and S. Martic), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany. doi: 10.1002/9783527683031.ch6
- ↑ Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG (1989). "A general method for site-specific incorporation of unnatural amino acids into proteins". Science. 244 (4901): 182–188. Bibcode:1989Sci...244..182N. doi:10.1126/science.2649980. PMID 2649980.
- ↑ Wang L, Xie J, Schultz PG (2006). "Expanding the genetic code". Annu Rev Biophys Biomol Struct. 35: 225–49. doi:10.1146/annurev.biophys.35.101105.121507. PMID 16689635.
- ↑ Sharma V, Wang Q, Lawrence DS (January 2008). "Peptide-based fluorescent sensors of protein kinase activity: design and applications". Biochim. Biophys. Acta. 1784 (1): 94–9. doi:10.1016/j.bbapap.2007.07.016. PMC 2684651. PMID 17881302.
- ↑ Violin JD, Zhang J, Tsein RY, Newton AC (2003). "A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C". J. Cell Biol. 161 (5): 899–909. doi:10.1083/jcb.200302125. PMC 2172956. PMID 12782683.
- ↑ Verveer PJ, Wouters FS, Hansra G, Bornancin F, Bastiaens PI (2000). "Quantitative imaging of lateral ERbB1 receptor signal propagation in the plasma membrane". Science. 290 (5496): 1567–1570. Bibcode:2000Sci...290.1567V. doi:10.1126/science.290.5496.1567. PMID 11090353.
- ↑ Muller S, Demotz S, Bulliard C, Valitutti S (1999). "Kinetics and extent of protein tyrosine kinase activation in individual T cells upon antigenic stimulation". Immunology. 97 (2): 287–293. doi:10.1046/j.1365-2567.1999.00767.x. PMC 2326824. PMID 10447744.
- 1 2 3 4 5 Che CM, Siu FM (2010). "Metal complexes in medicine with a focus on enzyme inhibition". Current Opinion in Chemical Biology. 14 (2): 255–261. doi:10.1016/j.cbpa.2009.11.015. PMID 20018553.
- ↑ Feazell RP, Nakayama-Ratchford N, Dai H, Lippard SJ (2007). "Soluble Single-Walled Carbon Nanotubes as Longboat Delivery Systems for Platinum(IV) Anticancer Drug Design". Journal of the American Chemical Society. 129 (27): 8438–9. doi:10.1021/ja073231f. PMC 2505197. PMID 17569542.
- ↑ Yang Z, Wang X, Diao H, Zhang J, Li H, Sun H, Guo Z (September 2007). "Encapsulation of platinum anticancer drugs by apoferritin". Chem. Commun. (Camb.) (33): 3453–5. doi:10.1039/b705326f. PMID 17700879.
- ↑ Angelucci F, Sayed AA, Williams DL, Boumis G, Brunori M, Dimastrogiovanni D, Miele AE, Pauly F, Bellelli A (2009). "Inhibition of Schistosoma mansoni Thioredoxin-glutathione Reductase by Auranofin: STRUCTURAL AND KINETIC ASPECTS". Journal of Biological Chemistry. 284 (42): 28977–85. doi:10.1074/jbc.M109.020701. PMC 2781444. PMID 19710012.
- 1 2 Lo YC, Ko TP, Su WC, Su TL, Wang AHJ (2009). "Terpyridine-platinum(II) complexes are effective inhibitors of mammalian topoisomerases and human thioredoxin reductase 1". Journal of Inorganic Biochemistry. 103 (7): 1082–1092. doi:10.1016/j.jinorgbio.2009.05.006. PMID 19525010.
- ↑ Becker K, Herold-Mende C, Park JJ, Lowe G, Schirmer RH (2001). "Human Thioredoxin Reductase is Efficiently Inhibited by (2,2':6',2' '-Terpyridine)platinum(II) Complexes. Possible Implications for a Novel Antitumor Strategy". Journal of Medicinal Chemistry. 44 (17): 2784–92. doi:10.1021/jm001014i. PMID 11495589.
- ↑ Arnesano F, Boccarelli A, Cornacchia D, Nushi F, Sasanelli R, Coluccia M, Natile G (2009). "Mechanistic Insight into the Inhibition of Matrix Metalloproteinases by Platinum Substrates†". Journal of Medicinal Chemistry. 52 (23): 7847–55. doi:10.1021/jm900845t. PMID 19757821.
- ↑ Ang WH, Parker LJ, De Luca A, Juillerat-Jeanneret L, Morton CJ, Lo Bello M, Parker MW, Dyson PJ (2009). "Rational design of an organometallic glutathione transferase inhibitor". Angew. Chem. Int. Ed. Engl. 48 (21): 3854–7. doi:10.1002/anie.200900185. PMID 19396894.
- ↑ Sun RW, Ma DL, Wong EL, Che CM (November 2007). "Some uses of transition metal complexes as anti-cancer and anti-HIV agents". Dalton Trans (43): 4884–92. doi:10.1039/b705079h. PMID 17992273.
- ↑ Hughes B (2009). "2008 FDA drug approvals". Nature Reviews Drug Discovery. 8 (2): 93–6. doi:10.1038/nrd2813. PMID 19180096.
- ↑ Benner SA, Sismour AM (2005). "Synthetic biology". Nature Reviews Genetics. 6 (7): 533–43. doi:10.1038/nrg1637. PMID 15995697.
- ↑ Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang R-Y, Algire MA, Benders GA, Montague MG, Ma L (2010). "Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome". Science. 329 (5987): 52–56. Bibcode:2010Sci...329...52G. doi:10.1126/science.1190719. PMID 20488990.
- ↑ Sismour AM, Lutz S, Park JH, Lutz MJ, Boyer PL, Hughes SH, Benner SA (2004). "PCR amplification of DNA containing non-standard base pairs by variants of reverse transcriptase from Human Immunodeficiency Virus-1". Nucleic Acids Research. 32 (2): 728–35. doi:10.1093/nar/gkh241. PMC 373358. PMID 14757837.
- 1 2 Kennan AJ, Haridas V, Severin K, Lee DH, Ghadiri MR (2001). "A de novo designed peptide ligase: A mechanistic investigation". J. Am. Chem. Soc. 123 (9): 1797–1803. doi:10.1021/ja991266c. PMID 11456796.
- ↑ Saghatelian A, Yokobayashi Y, Soltani K, Ghadiri MR (2001). "A chiroselective peptide replicator". Nature. 409 (6822): 797–801. Bibcode:2001Natur.409..797S. doi:10.1038/35057238. PMID 11236988.
- ↑ Martin VJJ, Pitera DJ, Withers ST, Newman JD, Keasling JD (2003). "Engineering a mevalonate pathway in Escherichia coli for production of terpenoids". Nature Biotechnology. 21 (7): 796–802. doi:10.1038/nbt833. PMID 12778056.
- ↑ Park SH, Zarrinpar A, Lim WA (2003). "Rewiring MAP kinase pathways using alternative scaffold assembly mechanisms". Science. 299 (5609): 1061–1064. Bibcode:2003Sci...299.1061P. doi:10.1126/science.1076979. PMID 12511654.
- ↑ Prehoda KE, Scott JA, Mullins RD, Lim WA (2000). "Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex". Science. 290 (5492): 801–806. Bibcode:2000Sci...290..801P. doi:10.1126/science.290.5492.801. PMID 11052943.
- ↑ Emre N, Coleman R, Ding S (June 2007). "A chemical approach to stem cell biology". Curr Opin Chem Biol. 11 (3): 252–8. doi:10.1016/j.cbpa.2007.04.024. PMID 17493865.
- ↑ Vazin T, Freed WJ (2010). "Human embryonic stem cells: derivation, culture, and differentiation: a review". Restor. Neurol. Neurosci. 28 (4): 589–603. doi:10.3233/RNN-2010-0543. PMC 2973558. PMID 20714081.
- ↑ Chen S, Do JT, Zhang Q, Yao S, Yan F, Peters EC, Schöler HR, Schultz PG, Ding S (November 2006). "Self-renewal of embryonic stem cells by a small molecule". Proc. Natl. Acad. Sci. USA. 103 (46): 17266–71. Bibcode:2006PNAS..10317266C. doi:10.1073/pnas.0608156103. PMC 1859921. PMID 17088537.
- ↑ Warashina M, Min KH, Kuwabara T, Huynh A, Gage FH, Schultz PG, Ding S (January 2006). "A synthetic small molecule that induces neuronal differentiation of adult hippocampal neural progenitor cells". Angew. Chem. Int. Ed. Engl. 45 (4): 591–3. doi:10.1002/anie.200503089. PMID 16323231.
- ↑ Egli D, Rosains J, Birkhoff G, Eggan K (2007). "Developmental reprogramming after chromosome transfer into mitotic mouse zygotes". Nature. 447 (7145): 679–85. Bibcode:2007Natur.447..679E. doi:10.1038/nature05879. PMID 17554301.
- ↑ Takahashi K, Yamanaka S (2006). "Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors". Cell. 126 (4): 663–76. doi:10.1016/j.cell.2006.07.024. PMID 16904174.
- ↑ Anastasia L, Pelissero G, Venerando B, Tettamanti G (2010). "Cell reprogramming: Expectations and challenges for chemistry in stem cell biology and regenerative medicine". Cell Death and Differentiation. 17 (8): 1230–7. doi:10.1038/cdd.2010.14. PMID 20168332.
- ↑ Chen S, Zhang Q, Wu X, Schultz PG, Ding S (2004). "Dedifferentiation of Lineage-Committed Cells by a Small Molecule". Journal of the American Chemical Society. 126 (2): 410–1. doi:10.1021/ja037390k. PMID 14719906.
- ↑ Chen S, Takanashi S, Zhang Q, Xiong W, Zhu S, Peters EC, Ding S, Schultz PG (June 2007). "Reversine increases the plasticity of lineage-committed mammalian cells". Proc. Natl. Acad. Sci. USA. 104 (25): 10482–7. Bibcode:2007PNAS..10410482C. doi:10.1073/pnas.0704360104. PMC 1965539. PMID 17566101.
- 1 2 3 Giepmans BNG, Adams SR, Ellisman MH, Tsien RY (2006). "The Fluorescent Toolbox for Assessing Protein Location and Function". Science. 312 (5771): 217–224. Bibcode:2006Sci...312..217G. doi:10.1126/science.1124618. PMID 16614209.
- ↑ Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS (2005). "Quantum Dots for Live Cells, in Vivo Imaging, and Diagnostics". Science. 307 (5709): 538–544. Bibcode:2005Sci...307..538M. doi:10.1126/science.1104274. PMC 1201471. PMID 15681376.
- ↑ Dave SR, Gao X (2009). "Monodisperse magnetic nanoparticles for biodetection, imaging, and drug delivery: a versatile and evolving technology". Wiley Interdiscip Rev Nanomed Nanobiotechnol. 1 (6): 583–609. doi:10.1002/wnan.51. PMID 20049819.
- ↑ Zinchuk V, Grossenbacher-Zinchuk O (2009). "Recent advances in quantitative colocalization analysis: Focus on neuroscience". Progress in Histochemistry and Cytochemistry. 44 (3): 125–72. doi:10.1016/j.proghi.2009.03.001. PMID 19822255.
- ↑ Galperin E, Verkhusha VV, Sorkin A (2004). "Three-chromophore FRET microscopy to analyze multiprotein interactions in living cells". Nature Methods. 1 (3): 209–17. doi:10.1038/nmeth720. PMID 15782196.
- ↑ Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH (2002). "Multicolor and Electron Microscopic Imaging of Connexin Trafficking". Science. 296 (5567): 503–507. Bibcode:2002Sci...296..503G. doi:10.1126/science.1068793. PMID 11964472.
- ↑ Terai T, Nagano T (2008). "Fluorescent probes for bioimaging applications". Current Opinion in Chemical Biology. 12 (5): 515–21. doi:10.1016/j.cbpa.2008.08.007. PMID 18771748.
- 1 2 Stoughton RB (2005). "Applications of DNA microarrays in biology". Annual Review of Biochemistry. 74: 53–82. doi:10.1146/annurev.biochem.74.082803.133212. PMID 15952881.
- 1 2 Southern, E. M. (2001) DNA Microarrays, pp 1–15.
- ↑ Lausted C, Dahl T, Warren C, King K, Smith K, Johnson M, Saleem R, Aitchison J, Hood L (2004). "POSaM: A fast, flexible, open-source, inkjet oligonucleotide synthesizer and microarrayer". Genome Biology. 5 (8): R58. doi:10.1186/gb-2004-5-8-r58. PMC 507883. PMID 15287980.
- ↑ Ahrendt SA, Halachmi S, Chow JT, Wu L, Halachmi N, Yang SC, Wehage S, Jen J, Sidransky D (June 1999). "Rapid p53 sequence analysis in primary lung cancer using an oligonucleotide probe array". Proc. Natl. Acad. Sci. USA. 96 (13): 7382–7. Bibcode:1999PNAS...96.7382A. doi:10.1073/pnas.96.13.7382. PMC 22094. PMID 10377423.
- ↑ Morozova O, Marra MA (2008). "Applications of next-generation sequencing technologies in functional genomics". Genomics. 92 (5): 255–64. doi:10.1016/j.ygeno.2008.07.001. PMID 18703132.
- ↑ Roth A, Gill R, Certa U (2003). "Temporal and spatial gene expression patterns after experimental stroke in a rat model and characterization of PC4, a potential regulator of transcription". Molecular and Cellular Neuroscience. 22 (3): 353–64. doi:10.1016/S1044-7431(02)00039-8. PMID 12691737.
- 1 2 Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R (2001). "Integrated Genomic and Proteomic Analyses of a Systematically Perturbed Metabolic Network". Science. 292 (5518): 929–934. Bibcode:2001Sci...292..929I. doi:10.1126/science.292.5518.929. PMID 11340206.
- ↑ Timperio AM, d'Alessandro A, Pariset L, d'Amici GM, Valentini A, Zolla L (2009). "Comparative proteomics and transcriptomics analyses of livers from two different Bos taurus breeds: "Chianina and Holstein Friesian"". Journal of Proteomics. 73 (2): 309–22. doi:10.1016/j.jprot.2009.09.015. PMID 19782776.
- ↑ Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, Buttarazzi MS, Sanofsky BA, McNamara KL (2010). "Integrative Genomic and Proteomic Analyses Identify Targets for Lkb1-Deficient Metastatic Lung Tumors". Cancer Cell. 17 (6): 547–59. doi:10.1016/j.ccr.2010.04.026. PMC 2901842. PMID 20541700.
- ↑ Klüsener S, Hacker S, Tsai YL, Bandow JE, Gust R, Lai EM, Narberhaus F (June 2010). "Proteomic and transcriptomic characterization of a virulence-deficient phosphatidylcholine-negative Agrobacterium tumefaciens mutant". Mol. Genet. Genomics. 283 (6): 575–89. doi:10.1007/s00438-010-0542-7. PMID 20437057.
- ↑ Piruzian E, Bruskin S, Ishkin A, Abdeev R, Moshkovskii S, Melnik S, Nikolsky Y, Nikolskaya T (2010). "Integrated network analysis of transcriptomic and proteomic data in psoriasis". BMC Systems Biology. 4: 41. doi:10.1186/1752-0509-4-41. PMC 2873316. PMID 20377895.
- ↑ Li B, Pattenden SG, Lee D, Gutiérrez J, Chen J, Seidel C, Gerton J, Workman JL (December 2005). "Preferential occupancy of histone variant H2AZ at inactive promoters influences local histone modifications and chromatin remodeling". Proc. Natl. Acad. Sci. USA. 102 (51): 18385–90. Bibcode:2005PNAS..10218385L. doi:10.1073/pnas.0507975102. PMC 1317944. PMID 16344463.
- ↑ Wade JT, Hall DB, Struhl K (2004). "The transcription factor Ifh1 is a key regulator of yeast ribosomal protein genes". Nature. 432 (7020): 1054–1058. Bibcode:2004Natur.432.1054W. doi:10.1038/nature03175. PMID 15616568.
- ↑ Mooney RA, Davis SE, Peters JM, Rowland JL, Ansari AZ, Landick R (2009). "Regulator Trafficking on Bacterial Transcription Units in Vivo". Molecular Cell. 33 (1): 97–108. doi:10.1016/j.molcel.2008.12.021. PMC 2747249. PMID 19150431.
- ↑ Zeitlinger J, Simon I, Harbison CT, Hannett NM, Volkert TL, Fink GR, Young RA (2003). "Program-Specific Distribution of a Transcription Factor Dependent on Partner Transcription Factor and MAPK Signaling". Cell. 113 (3): 395–404. doi:10.1016/S0092-8674(03)00301-5. PMID 12732146.
- ↑ Farnham PJ (2009). "Insights from genomic profiling of transcription factors". Nature Reviews Genetics. 10 (9): 605–16. doi:10.1038/nrg2636. PMC 2846386. PMID 19668247.
- ↑ Lee HJ, Goodrich TT, Corn RM (2001). "SPR Imaging Measurements of 1-D and 2-D DNA Microarrays Created from Microfluidic Channels on Gold Thin Films". Analytical Chemistry. 73 (22): 5525–31. doi:10.1021/ac010762s. PMID 11816583.
- ↑ Makowska-Grzyska M, Kaguni JM (2010). "Primase Directs the Release of DnaC from DnaB". Molecular Cell. 37 (1): 90–101. doi:10.1016/j.molcel.2009.12.031. PMC 2819048. PMID 20129058.
- ↑ Bunnage ME, et al. (2015). "Know your target, know your molecule.". Nat. Chem. Biol. 1: 368–372. doi:10.1038/nchembio.1813.
- ↑ Huber KV, et al. (2014). "Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy.". Nature. 508: 222–227. doi:10.1038/nature13194.
- ↑ Chung C-W, et al. (2011). "Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains.". J. Med. Chem. 54: 3827–3838. doi:10.1021/jm200108t.
- ↑ Weygant N, et al. (2014). "Small molecule kinase inhibitor LRRK2-IN-1 demonstrates potent activity against colorectal and pancreatic cancer through inhibition of doublecortin-like kinase 1.". Mol. Cancer. 13: 103. doi:10.1186/1476-4598-13-103.
- ↑ Fox D, Burgin AB, Gurney ME (2014). "Structural basis for the design of selective phosphodiesterase 4B inhibitors.". Cell. Signaling. 26: 657–663. doi:10.1016/j.cellsig.2013.12.003.
- ↑ Tate EW, Kalesh KA, Lanyon-Hogg T, Storck EM, Thinon E (2015). "Global profiling of protein lipidation using chemical proteomic technologies.". Curr. Opin. Chem. Biol. 24: 48–57. doi:10.1016/j.cbpa.2014.10.016.
- ↑ Paulsen CE, Carroll KS (2013). "Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery.". Chem. Rev. 113: 4633–4679. doi:10.1021/cr300163e.
- ↑ Honigberg LA, et al. (2010). "The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy.". Proc. Natl. Acad. Sci. USA. 107: 13075–13080. doi:10.1073/pnas.1004594107.
- ↑ Padron D, et al. (2007). "Epidermal growth factor receptors with tyrosine kinase domain mutations exhibit reduced Cbl association, poor ubiquitylation, and down-regulation but are efficiently internalized.". Cancer Research. 67: 7695–7702. doi:10.1158/0008-5472.can-07-0484.
- ↑ Neklesa TK, Crews CM (2012). "Chemical biology: greasy tags for protein removal.". Nature. 487: 308–309. doi:10.1038/487308a.
- ↑ Patricelli MP, et al. (2006). "Functional Interrogation of the Kinome Using Nucleotide Acyl Phosphates.". Biochemistry. 46: 350–358. doi:10.1021/bi062142x.
- ↑ Lanning BR, et al. (2014). "A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors.". Nat. Chem. Biol. 10: 760–767. doi:10.1038/nchembio.158.
- ↑ Bantscheff M, et al. "Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes". Nat. Biotech. 29: 255–265. doi:10.1038/nbt.1759.
- ↑ Savitski MM, et al. (2014). "Tracking cancer drugs in living cells by thermal profiling of the proteome.". Science. 346: 6205. doi:10.1126/science.1255784.
- ↑ Ito T, et al. (2010). "Identification of a Primary Target of Thalidomide Teratogenicity.". Science. 327: 1345–1350. doi:10.1126/science.1177319.
- ↑ Lomernick B, et al. (2009). "Target identification using drug affinity responsive target stability (DARTS).". Proc. Natl. Acad. Sci. USA. 106: 21984–21989. doi:10.1073/pnas.0910040106.
- ↑ West GM, et al. (2010). "Quantitative proteomics approach for identifying protein–drug interactions in complex mixtures using protein stability measurements". Proc. Natl. Acad. Sci. USA. 107: 9078–9082. doi:10.1073/pnas.1000148107.
- ↑ Winter GE, et al. (2014). "The solute carrier SLC35F2 enables YM155-mediated DNA damage toxicity.". Nat. Chem. Biol. 10: 768–773. doi:10.1038/nchembio.1590.
- ↑ Hatzivassiliou G, et al. (2010). "RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth.". Nature. 464: 431–435. doi:10.1038/nature08833.
- ↑ Ahn K, et al. (2011). "Mechanistic and Pharmacological Characterization of PF-04457845: A Highly Potent and Selective Fatty Acid Amide Hydrolase Inhibitor That Reduces Inflammatory and Noninflammatory Pain". J. Pharmacol. Exp. Ther. 338: 114–124. doi:10.1124/jpet.111.180257.
- ↑ Hett EC, et al. "Rational targeting of active site tyrosine residues using sulfonyl fluoride probes". ACS Chem. Biol. 10: 1094–1098. doi:10.1021/cb5009475.
Further reading
- Dertinger SKW, Chiu DT, Jeon NL, Whitesides GM (2001). "Generation of gradients having complex shapes using microfluidic networks". Analytical Chemistry. 73: 1240–1246. doi:10.1021/ac001132d.
- Greif D, Pobigaylo N, Frage B, Becker A, Regtmeier J, Anselmetti D (2010). "Space- and time-resolved protein dynamics in single bacterial cells observed on a chip". Journal of Biotechnology. 149 (4): 280–288. doi:10.1016/j.jbiotec.2010.06.003. PMID 20599571.
- Li L, Ismagilov RF (2010). "Protein crystallization using microfluidic technologies based on valves, droplets, and SlipChip". Annu Rev Biophys. 39: 139–58. doi:10.1146/annurev.biophys.050708.133630. PMID 20192773.
- Lucchetta EM, Lee JH, Fu LA, Patel NH, Ismagilov RF (2005). "Dynamics of Drosophila embryonic patterning network perturbed in space and time using microfluidics". Nature. 434 (7037): 1134–1138. Bibcode:2005Natur.434.1134L. doi:10.1038/nature03509. PMC 2656922. PMID 15858575.
- Melin J, Quake SR (2007). "Microfluidic large-scale integration: The evolution of design rules for biological automation". Annual Review of Biophysics and Biomolecular Structure. 36: 213–231. doi:10.1146/annurev.biophys.36.040306.132646. PMID 17269901.
- Shen F, Du WB, Kreutz JE, Fok A, Ismagilov RF (2010). "Digital PCR on a SlipChip". Lab on a Chip. 10 (20): 2666–2672. doi:10.1039/c004521g. PMC 2948063. PMID 20596567.
- Song H, Chen DL, Ismagilov RF (2006). "Reactions in droplets in microflulidic channels". Angewandte Chemie International Edition. 45: 7336–7356. doi:10.1002/anie.200601554. PMC 1766322. PMID 17086584.
- Spiller DG, Wood CD, Rand DA, White MRH (2010). "Measurement of single-cell dynamics". Nature. 465 (7299): 736–745. Bibcode:2010Natur.465..736S. doi:10.1038/nature09232. PMID 20535203.
- Tice JD, Song H, Lyon AD, Ismagilov RF (2003). "Formation of droplets and mixing in multiphase microfluidics at low values of the Reynolds and the capillary numbers". Langmuir. 19 (22): 9127–9133. doi:10.1021/la030090w.
- Vincent ME, Liu WS, Haney EB, Ismagilov RF (2010). "Microfluidic stochastic confinement enhances analysis of rare cells by isolating cells and creating high density environments for control of diffusible signals". Chemical Society Reviews. 39 (3): 974–984. doi:10.1039/b917851a. PMC 2829723. PMID 20179819.
- Weibel DB, Whitesides GM (2006). "Applications of microfluidics in chemical biology". Current Opinion in Chemical Biology. 10 (6): 584–591. doi:10.1016/j.cbpa.2006.10.016. PMID 17056296.
- Whitesides GM (2006). "The origins and the future of microfluidics". Nature. 442 (7101): 368–373. Bibcode:2006Natur.442..368W. doi:10.1038/nature05058. PMID 16871203.
- Young EWK, Beebe DJ (2010). "Fundamentals of microfluidic cell culture in controlled microenvironments". Chemical Society Reviews. 39 (3): 1036–1048. doi:10.1039/b909900j. PMC 2967183. PMID 20179823.
Journals
- ACS Chemical Biology – The new Chemical Biology journal from the American Chemical Society.
- Bioorganic & Medicinal Chemistry – The Tetrahedron Journal for Research at the Interface of Chemistry and Biology
- ChemBioChem – A European Journal of Chemical Biology
- Chemical Biology – A point of access to chemical biology news and research from across RSC Publishing
- Chemistry & Biology – An interdisciplinary journal that publishes papers of exceptional interest in all areas at the interface between chemistry and biology. chembiol.com
- Journal of Chemical Biology – A new journal publishing novel work and reviews at the interface between biology and the physical sciences, published by Springer. link
- Journal of the Royal Society Interface – A cross-disciplinary publication promoting research at the interface between the physical and life sciences
- Molecular BioSystems – Chemical biology journal with a particular focus on the interface between chemistry and the -omic sciences and systems biology.
- Nature Chemical Biology – A monthly multidisciplinary journal providing an international forum for the timely publication of significant new research at the interface between chemistry and biology.
- Wiley Encyclopedia of Chemical Biology link