Microbiota
A microbiota is "the ecological community of commensal, symbiotic and pathogenic microorganisms that literally share our body space".[1][2] Joshua Lederberg coined the term, emphasising the importance of microorganisms inhabiting the human body in health and disease. Many scientific articles distinguish microbiome and microbiota to describe either the collective genomes of the microorganisms that reside in an environmental niche or the microorganisms themselves, respectively.[3][4][5] However, by the original definitions, these terms are largely synonymous.
The microbes being discussed generally do not cause disease unless they grow abnormally nonpathogenic organisms; they exist in harmony and symbiotically with their hosts.[6] The microbiome and host may have emerged as a unit by the process of integration.[7]
Introduction
All plants and animals, from protists to humans, live in close association with microbial organisms (see for example the human microbiome). Up until relatively recently, however, biologists have defined the interactions of plants and animals with the microbial world mostly in the context of disease states and of a relatively small number of symbiotic case studies. Organisms do not live in isolation, but have evolved in the context of complex communities. A number of advances have driven a change in the perception of microbiomes, including:
- the ability to perform genomic and gene expression analyses of single cells and even of entire microbial communities in the new disciplines of metagenomics and metatranscriptomics
- massive databases making this information accessible to researchers across multiple disciplines
- methods of mathematical analysis that help researchers to make sense of complex data sets
Increasingly, biologists have come to appreciate that microbes make up an important part of an organism's phenotype, far beyond the occasional symbiotic case study.[8]
Pierre-Joseph van Beneden (1809-1894), a Belgian professor at the University of Louvain, developed the concept of commensalism during the nineteenth century. In his 1875 publication Animal Parasites and Messmates, Van Beneden presented 264 examples of commensalism. His conception was widely accepted by his contemporaries and commensalism has continued to be used as a concept right up to the present day: microbiome is clearly linked to commensalism.[9]
Microbiota by host
There is a strengthening consensus among evolutionary biologists that one should not separate an organism's genes from the context of its resident microbes.
Humans
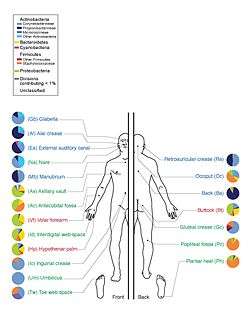
The human microbiota includes bacteria, fungi, and archaea. Micro-animals which live on the human body are excluded. The human microbiome refers to their genomes.[10]
Humans are colonized by many microorganisms; the traditional estimate was that humans live with ten times more non-human cells than human cells; more recent estimates have lowered that to 3:1 and even to approximately the same number; all the numbers are estimates.[11][12][13][14] Regardless of the exact number, the microbiota that colonize humans have not merely a commensal (a non-harmful coexistence), but rather a mutualistic relationship with their human hosts.[10]:700[15] Some of these organisms perform tasks that are known to be useful for the human host; for most, the role is not well understood. Those that are expected to be present, and that under normal circumstances do not cause disease, are deemed normal flora or normal microbiota.[10]
The Human Microbiome Project took on the project of sequencing the genome of the human microbiota, focusing particularly on the microbiota that normally inhabit the skin, mouth, nose, digestive tract, and vagina.[10] It reached a milestone in 2012 when it published initial results.[16]
Non-human animals

- A massive, worldwide decline in amphibian populations has been well-publicised. Habitat loss and over-exploitation account for part of the problem, but many other processes seem to be at work. The spread of the virulent fungal disease chytridiomycosis represents an enigma.[17] The ability of some species to coexist with the causative agent Batrachochytrium dendrobatidis appears to be due to the expression of antimicrobial skin peptides along with the presence of symbiotic microbes that benefit the host by resisting pathogen colonization or inhibiting their growth while being themselves resistant to high concentrations of antimicrobial skin peptides.[18]
- The bovine rumen harbors a complex microbiome that converts plant cell wall biomass into proteins, short chain fatty acids, and gases. Multiple species are involved in this conversion. Traditional methods of characterizing the microbial population, based on culture analysis, missed many of the participants in this process. Comparative metagenomic studies yielded the surprising result that individual steer had markedly different community structures, predicted phenotype, and metabolic potentials,[19] even though they were fed identical diets, were housed together, and were apparently functionally identical in their utilization of plant cell wall resources.
- Leaf-cutter ants form huge underground colonies with millions of workers, each colony harvesting hundreds of kilograms of leaves each year. Unable to digest the cellulose in the leaves directly, they maintain fungus gardens that are the colony's primary food source. The fungus itself does not digest cellulose. Instead, a microbial community containing a diversity of bacteria is responsible for cellulose digestion. Analysis of the microbial population's genomic content by community metagenome sequencing methods revealed the presence of many genes with a role in cellulose digestion. This microbiome's predicted carbohydrate-degrading enzyme profile is similar to that of the bovine rumen, but the species composition is almost entirely different.[20]
- Mice are the most used models for human disease. As more and more diseases are linked to dysfunctional microbiomes, mice have become the most studied organism in this regard. Mostly it is the gut microbiota that have been studied in relation to allergic airway disease, obesity, gastrointestinal diseases and diabetes. Intriguingly, recent work has shown that perinatal shifting of microbiota through administration of low dose antibiotics can have long-lasting effects on future susceptibility to allergic airway disease.[21][22] These studies showed a remarkable link between the frequency of certain subsets of microbes and disease severity. In aggregate these studies suggest that the presence of specific microbes, early in postnatal life, play an instructive role in the development of future immune responses. Mechanistically, a recent study done on gnotobiotic mice described a method in which certain strains of gut bacteria were found to transmit a particular phenotype to recipient germ-free mice, identifying an unanticipated range of bacterial strains that promoted accumulation of colonic regulatory T cells, as well as strains that modulated mouse adiposity and cecal metabolite concentrations. Another study showed that when adult germ-free mice were colonized with the gut flora of obese mice, there was a dramatic weight increase and an observed increased metabolism of monosaccharides and short-chain fatty acids. Looking at the gut flora compositions between normal and obese mice, obese mice had less Bacteroidetes than Firmicutes in abundance in gut flora and it is hypothesized that the microbiota of obese mice are more efficient at extracting energy from food.[23] This combinatorial approach enables a systems-level understanding of microbial contributions to human biology.[24] But also other mucoide tissues as lung and vagina have been studied in relation to diseases such as asthma, allergy and vaginosis [25]
Plants

- Plants exhibit a broad range of relationships with symbiotic microorganisms, ranging from parasitism, in which the association is disadvantageous to the host organism, to mutualism, in which the association is beneficial to both, to commensalism, in which the symbiont benefits while the host is not affected. Exchange of nutrients between symbiotic partners is an important part of the relationship: it may be bidirectional or unidirectional, and it may be context dependent. The strategies for nutrient exchange are highly diverse. Oomycetes and fungi have, through convergent evolution, developed similar morphology and occupy similar ecological niches. They develop hyphae, filamentous structures that penetrate the host cell. In those cases where the association is mutualistic, the plant often exchanges hexose sugars for inorganic phosphate from the fungal symbiont. It is speculated that such associations, which are very ancient, may have aided plants when they first colonized land.[26][27]
- A huge range of bacterial symbionts colonize plants. Many of these are pathogenic, but others known as plant-growth promoting bacteria (PGPB) provide the host with essential services such as nitrogen fixation, solubilization of minerals such as phosphorus, synthesis of plant hormones, direct enhancement of mineral uptake, and protection from pathogens.[28][29] PGPBs may protect plants from pathogens by competing with the pathogen for an ecological niche or a substrate, producing inhibitory allelochemicals, or inducing systemic resistance in host plants to the pathogen[30]
- Plants are attractive hosts for microorganisms since they provide a variety of nutrients. Microorganisms on plants can be epiphytes (found on the plants) or endophytes (found inside plant tissue).[31][32]
Immune system
The symbiotic relationship between a mammalian host and its microbiota has a significant impact on shaping the host's immune system.[33] In many animals, the immune system and microbiota engage in "cross-talk", exchanging chemical signals. This allows the immune system to recognize the types of bacteria that are harmful to the host and combat them, while allowing the helpful bacteria to carry out their functions; in turn, the microbiota influence immune reactivity and targeting.[34] Bacteria can be transferred from mother to child through direct contact and after birth, or through indirect contact through eggs, coprophagy, and several other pathways.[35] As the infant microbiome is established, commensal bacteria quickly populate the gut, prompting a range of immune responses and "programming" the immune system with long-lasting effects.[34] This early colonization helps to establish the symbiotic microbiome inside the animal host early in its life.[33] The bacteria are also able to stimulate lymphoid tissue associated with the gut mucosa. This enables the tissue to produce antibodies for pathogens that may enter the gut.
It has been found that bacteria may also play a role in the activation of TLRs (toll-like receptors) in the intestines. TLRs are a type of PRR (pattern recognition receptor) used by host cells to help repair damage and recognize dangers to the host. This could be important in immune tolerance and autoimmune diseases. Pathogens could influence this symbiotic coexistence leading to immune dysregulation and susceptibility to diseases. This could provide new direction for managing immunological and metabolic diseases.[36]
Co-evolution of microbiota

Organisms evolve within eco-systems so that the change of one organism affects the change of others. Co-evolution (also called "hologenome theory") proposes that an object of natural selection is not the individual organism, but the organism together with its associated organisms, includings its microbial communities.
Coral reefs. The hologenome theory originated in studies on coral reefs. Coral reefs are the largest structures created by living organisms, and contain abundant and highly complex microbial communities. Over the past several decades, major declines in coral populations have occurred. Climate change, water pollution and over-fishing are three stress factors that have been described as leading to disease susceptibility. Over twenty different coral diseases have been described, but of these, only a handful have had their causative agents isolated and characterized. Coral bleaching is the most serious of these diseases. In the Mediterranean Sea, the bleaching of Oculina patagonica was first described in 1994 and shortly determined to be due to infection by Vibrio shiloi. From 1994 to 2002, bacterial bleaching of O. patagonica occurred every summer in the eastern Mediterranean. Surprisingly, however, after 2003, O. patagonica in the eastern Mediterranean has been resistant to V. shiloi infection, although other diseases still cause bleaching. The surprise stems from the knowledge that corals are long lived, with lifespans on the order of decades,[37] and do not have adaptive immune systems. Their innate immune systems do not produce antibodies, and they should seemingly not be able to respond to new challenges except over evolutionary time scales. The puzzle of how corals managed to acquire resistance to a specific pathogen led Eugene Rosenberg and Ilana Zilber-Rosenberg to propose the Coral Probiotic Hypothesis. This hypothesis proposes that a dynamic relationship exists between corals and their symbiotic microbial communities. By altering its composition, this holobiont can adapt to changing environmental conditions far more rapidly than by genetic mutation and selection alone. Extrapolating this hypothesis of adaptation and evolution to other organisms, including higher plants and animals, led to the proposal of the Hologenome Theory of Evolution.[38]
The hologenome theory is still being debated.[39] A major criticism has been the claim that V. shiloi was misidentified as the causative agent of coral bleaching, and that its presence in bleached O. patagonica was simply that of opportunistic colonization.[40] If this is true, the basic observation leading to the theory would be invalid. Nevertheless, the theory has gained significant popularity as a way of explaining rapid changes in adaptation that cannot otherwise be explained by traditional mechanisms of natural selection. For those who accept the hologenome theory, the holobiont has become the principal unit of natural selection. On the other hand, it has been stated that the holobiont is the result of other step of integration that it is also observed at the cell (symbiogenesis, endosymbiosis) and genomic levels.[7]
Research methods
Targeted amplicon sequencing
Targeted amplicon sequencing relies on having some expectations about the composition of the community that is being studied. In target amplicon sequencing a phylogenetically informative marker is targeted for sequencing. Such a marker should be present in ideally all the expected organisms. It should also evolve in such a way that it is conserved enough that primers can target genes from a wide range of organisms while evolving quickly enough to allow for finer resolution at the taxonomic level. A common marker for human microbiome studies is the gene for bacterial 16S rRNA (i.e. "16S rDNA", the sequence of DNA which encodes the ribosomal RNA molecule).[41] Since ribosomes are present in all living organisms, using 16S rDNA allows for DNA to be amplified from many more organisms than if another marker were used. The 16S rDNA gene contains both slowly evolving regions and fast evolving regions; the former can be used to design broad primers while the latter allow for finer taxonomic distinction. However, species-level resolution is not typically possible using the 16S rDNA. Primer selection is an important step, as anything that cannot be targeted by the primer will not be amplified and thus will not be detected. Different sets of primers have been shown to amplify different taxonomic groups due to sequence variation.
Targeted studies of eukaryotic and viral communities are limited[42] and subject to the challenge of excluding host DNA from amplification and the reduced eukaryotic and viral biomass in the human microbiome.[43]
After the amplicons are sequenced, molecular phylogenetic methods are used to infer the composition of the microbial community. This is done by clustering the amplicons into operational taxonomic units (OTUs) and inferring phylogenetic relationships between the sequences. Due to the complexity of the data, distance measures such as UniFrac distances are usually defined between microbiome samples, and downstream multivariate methods are carried out on the distance matrices. An important point is that the scale of data is extensive, and further approaches must be taken to identify patterns from the available information. Tools used to analyze the data include VAMPS,[44] QIIME[45] and mothur.[46]
Metagenomic sequencing
Metagenomics is also used extensively for studying microbial communities.[47][48][49] In metagenomic sequencing, DNA is recovered directly from environmental samples in an untargeted manner with the goal of obtaining an unbiased sample from all genes of all members of the community. Recent studies use shotgun Sanger sequencing or pyrosequencing to recover the sequences of the reads.[50] The reads can then be assembled into contigs. To determine the phylogenetic identity of a sequence, it is compared to available full genome sequences using methods such as BLAST. One drawback of this approach is that many members of microbial communities do not have a representative sequenced genome.[41]
Despite the fact that metagenomics is limited by the availability of reference sequences, one significant advantage of metagenomics over targeted amplicon sequencing is that metagenomics data can elucidate the functional potential of the community DNA.[51][52] Targeted gene surveys cannot do this as they only reveal the phylogenetic relationship between the same gene from different organisms. Functional analysis is done by comparing the recovered sequences to databases of metagenomic annotations such as KEGG. The metabolic pathways that these genes are involved in can then be predicted with tools such as MG-RAST,[53] CAMERA[54] and IMG/M.[55]
RNA and protein-based approaches
Metatranscriptomics studies have been performed to study the gene expression of microbial communities through methods such as the pyrosequencing of extracted RNA.[56] Structure based studies have also identified non-coding RNAs (ncRNAs) such as ribozymes from microbiota.[57] Metaproteomics is a new approach that studies the proteins expressed by microbiota, giving insight into its functional potential.[58]
Projects
The Human Microbiome Project (HMP) was a United States National Institutes of Health initiative with the goal of identifying and characterizing the microorganisms which are found in association with both healthy and diseased humans (their microbial flora).[59] Launched in 2008, it was a five-year project, best characterized as a feasibility study, with a total budget of $115 million. The ultimate goal of this and similar NIH-sponsored microbiome projects is to test how changes in the human microbiome are associated with human health or disease.[59]
The Earth Microbiome Project (EMP) is an initiative to collect natural samples and analyze the microbial community around the globe. Microbes are highly abundant, diverse and have an important role in the ecological system. Yet as of 2010, it was estimated that the total global environmental DNA sequencing effort had produced less than 1 percent of the total DNA found in a liter of seawater or a gram of soil,[60] and the specific interactions between microbes are largely unknown. The EMP aims to process as many as 200,000 samples in different biomes, generating a complete database of microbes on earth to characterize environments and ecosystems by microbial composition and interaction. Using these data, new ecological and evolutionary theories can be proposed and tested.[61]
The Brazilian Microbiome Project (BMP) aims to assemble a Brazilian Microbiome Consortium/Database. At present, many metagenomic projects underway in Brazil are widely known. Our goal is to co-ordinate and standardize these, together with future projects. This is the first attempt to collect and collate information about Brazilian microbial genetic and functional diversity in a systematic and holistic manner. New sequence data have been generated from samples collected in all Brazilian regions, however the success of the BMP depends on a massive collaborative effort of both the Brazilian and international scientific communities. Therefore, we invite all colleagues to participate in this project. There is no prioritization of specific taxonomic groups, studies could include any ecosystem, and all proposals and any help will be very welcome.
Privacy
The DNA of the microbes that inhabit a person's human body can uniquely identify the person. A risk to violating a person's privacy may exist, if the person anonymously donated microbe DNA data, and the data could be used to identify the person and their medical condition, and if the person's identity were revealed.[62][63][64][65]
See also
Notes
References
- ↑ Lederberg, J; McCray, AT (2001). "'Ome Sweet 'Omics—a genealogical treasury of words". Scientist. 15: 8.
- ↑ Nih Hmp Working, Group; Peterson, J; Garges, S; Giovanni, M; McInnes, P; Wang, L; Schloss, J. A.; Bonazzi, V; McEwen, J. E.; Wetterstrand, K. A.; Deal, C; Baker, C. C.; Di Francesco, V; Howcroft, T. K.; Karp, R. W.; Lunsford, R. D.; Wellington, C. R.; Belachew, T; Wright, M; Giblin, C; David, H; Mills, M; Salomon, R; Mullins, C; Akolkar, B; Begg, L; Davis, C; Grandison, L; Humble, M; et al. (2009). "The NIH Human Microbiome Project". Genome Res. 19 (12): 2317–2323. doi:10.1101/gr.096651.109. PMC 2792171
 . PMID 19819907.
. PMID 19819907. - ↑ Backhed, F; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. (2005). "Host-Bacterial Mutualism in the Human Intestine". Science. 307 (5717): 1915–1920. Bibcode:2005Sci...307.1915B. doi:10.1126/science.1104816. PMID 15790844.
- ↑ Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. (2007). "The Human Microbiome Project". Nature. 449 (7164): 804–810. Bibcode:2007Natur.449..804T. doi:10.1038/nature06244. PMC 3709439. PMID 17943116.
- ↑ Ley, R.E.; Peterson, D.A.; Gordon, J.I. (2006). "Ecological and Evolutionary Forces Shaping Microbial Diversity in the Human Intestine". Cell. 124 (4): 837–848. doi:10.1016/j.cell.2006.02.017. PMID 16497592.
- ↑ Madigan, Michael T. (2012). Brock biology of microorganisms (13th ed.). San Francisco: Benjamin Cummings. ISBN 9780321649638.
- 1 2 Salvucci, E. (2014). "Microbiome, holobiont and the net of life". Critical Reviews in Microbiology: 1–10. doi:10.3109/1040841X.2014.962478.
- ↑ Bosch, T. C. G.; McFall-Ngai, M. J. (2011). "Metaorganisms as the new frontier". Zoology. 114 (4): 185–190. doi:10.1016/j.zool.2011.04.001. PMID 21737250.
- ↑ Poreau B., Biologie et complexité : histoire et modèles du commensalisme. PhD Dissertation, University of Lyon, France, 2014.
- 1 2 3 4 Sherwood, Linda; Willey, Joanne; Woolverton, Christopher (2013). Prescott's Microbiology (9th ed.). New York: McGraw Hill. pp. 713–721. ISBN 9780073402406. OCLC 886600661.
- ↑ American Academy of Microbiology FAQ: Human Microbiome January 2014
- ↑ Judah L. Rosner for Microbe Magazine, Feb 2014. Ten Times More Microbial Cells than Body Cells in Humans?
- ↑ Alison Abbott for Nature News. Jan 8 2016 Scientists bust myth that our bodies have more bacteria than human cells
- ↑ Sender, R; Fuchs, S; Milo, R (Jan 2016). "Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans". Cell. 164 (3): 337–40. doi:10.1016/j.cell.2016.01.013. PMID 26824647.
- ↑ Quigley, EM (Sep 2013). "Gut bacteria in health and disease". Gastroenterol Hepatol (N Y). 9 (9): 560–9. PMC 3983973. PMID 24729765.
- ↑ "NIH Human Microbiome Project defines normal bacterial makeup of the body". NIH News. 13 June 2012.
- ↑ Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues ASL, Fischman DL, Waller RW; Chanson; Cox; Young; Rodrigues; Fischman; Waller (2004). "Status and Trends of Amphibian Declines and Extinctions Worldwide" (PDF). Science. 306 (5702): 1783–6. Bibcode:2004Sci...306.1783S. doi:10.1126/science.1103538. PMID 15486254.
- ↑ Woodhams DC, Rollins-Smith LA, Alford RA, Simon MA, Harris RN (2007). "Innate immune defenses of amphibian skin: antimicrobial peptides and more". Animal Conservation. 10 (4): 425–8. doi:10.1111/j.1469-1795.2007.00150.x.
- ↑ Brulc JM, Antonopoulos DA, Miller MEB, Wilson MK, Yannarell AC, Dinsdale EA, Edwards RE, Frank ED, Emerson JB, Wacklin P, Coutinho PM, Henrissat B, Nelson KE, White BA; Antonopoulos; Berg Miller; Wilson; Yannarell; Dinsdale; Edwards; Frank; Emerson; Wacklin; Coutinho; Henrissat; Nelson; White (2009). "Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases". Proc. Natl. Acad. Sci. USA. 106 (6): 1948–53. Bibcode:2009PNAS..106.1948B. doi:10.1073/pnas.0806191105. PMC 2633212. PMID 19181843.
- ↑ Suen G, Scott JJ, Aylward FO, Adams SM, Tringe SG, Pinto-Tomás AA, Foster CE, Pauly M, Weimer PJ, Barry KW, Goodwin LA, Bouffard P, Li L, Osterberger J, Harkins TT, Slater SC, Donohue TJ, Currie CR (2010). Sonnenburg, Justin, ed. "An Insect Herbivore Microbiome with High Plant Biomass-Degrading Capacity". PLoS Genet. 6 (9): e1001129. doi:10.1371/journal.pgen.1001129. PMC 2944797. PMID 20885794.
- ↑ Russell SL, , Gold MJ; et al. (May 2012). "Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma". EMBO Rep. 13 (5): 440–7. doi:10.1038/embor.2012.32. PMC 3343350. PMID 22422004.
- ↑ Russell SL, Gold MJ, et al. (Aug 2014). "Perinatal antibiotic-induced shifts in gut microbiota have differential effects on inflammatory lung diseases". J Allergy Clin Immunol. 135 (1): 100–9. doi:10.1016/j.jaci.2014.06.027. PMID 25145536.
- ↑ Turnbaugh PJ, et al. (Dec 2006). "An obesity-associated gut microbiome with increased capacity for energy harvest" (PDF). Nature. 444 (7122): 1027–31. Bibcode:2006Natur.444.1027T. doi:10.1038/nature05414. PMID 17183312.
- ↑ Faith JJ, Ahern PP, Ridaura VK, et al. (Jan 2014). "Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice.". Sci Transl Med. 6 (220): 220. doi:10.1126/scitranslmed.3008051. PMC 3973144. PMID 24452263.
- ↑ Barfod, KK; Roggenbuck, M; Hansen, LH; Schjørring, S; Larsen, ST; Sørensen, SJ; Krogfelt, KA (2013). "The murine lung microbiome in relation to the intestinal and vaginal bacterial communities". BMC Microbiol. 13: 303. doi:10.1186/1471-2180-13-303.
- ↑ Remy W, Taylor TN, Hass H, Kerp H; Taylor; Hass; Kerp (1994). "Four hundred-million-year-old vesicular arbuscular mycorrhizae" (PDF). Proc. Natl. Acad. Sci. USA. 91 (25): 11841–3. Bibcode:1994PNAS...9111841R. doi:10.1073/pnas.91.25.11841. PMC 45331. PMID 11607500.
- ↑ Chibucos MC, Tyler BM (2009). "Common themes in nutrient acquisition by plant symbiotic microbes, described by the Gene Ontology". BMC Microbiology. 9(Suppl 1): S6. doi:10.1186/1471-2180-9-S1-S6.
- ↑ Kloepper, J. W (1993). "Plant growth-promoting rhizobacteria as biological control agents". In Metting, F. B. Jr. Soil microbial ecology: applications in agricultural and environmental management. New York: Marcel Dekker Inc. pp. 255–274. ISBN 0-8247-8737-4.
- ↑ Bloemberg, G. V.; Lugtenberg, B. J. J. (2001). "Molecular basis of plant growth promotion and biocontrol by rhizobacteria". Current Opinion in Plant Biology. 4 (4): 343–350. doi:10.1016/S1369-5266(00)00183-7. PMID 11418345.
- ↑ Compant S, Duffy B, Nowak J, Clément C, Barka EA (2005). "Use of Plant Growth-Promoting Bacteria for Biocontrol of Plant Diseases: Principles, Mechanisms of Action, and Future Prospects". Appl Environ Microbiol. 71 (9): 4951–9. doi:10.1128/AEM.71.9.4951-4959.2005. PMC 1214602. PMID 16151072.
- ↑ Berlec, Aleš (2012-09-01). "Novel techniques and findings in the study of plant microbiota: Search for plant probiotics". Plant Science. 193–194: 96–102. doi:10.1016/j.plantsci.2012.05.010. PMID 22794922.
- ↑ Whipps, J.m.; Hand, P.; Pink, D.; Bending, G.d. (2008-12-01). "Phyllosphere microbiology with special reference to diversity and plant genotype". Journal of Applied Microbiology. 105 (6): 1744–1755. doi:10.1111/j.1365-2672.2008.03906.x. ISSN 1365-2672. PMID 19120625.
- 1 2 Round, June L.; O'Connell, Ryan M.; Mazmanian, Sarkis K. (2010). "Coordination of tolerogenic immune responses by the commensal microbiota". Journal of Autoimmunity. 34 (3): J220–J225. doi:10.1016/j.jaut.2009.11.007. PMID 19963349.
- 1 2 Cahenzli, Julia; Balmer, Maria L.; McCoy, Kathy D. (2012). "Microbial-immune cross-talk and regulation of the immune system". Immunology. 138 (1): 12–22. doi:10.1111/j.1365-2567.2012.03624.x. PMC 3533697. PMID 22804726.
- ↑ Rosenberg, Eugene; Zilber-Rosenberg, Ilana (2016). "Microbes Drive Evolution of Animals and Plants: the Hologenome Concept" (PDF). MBio. 7 (2): e01395–15. doi:10.1128/mbio.01395-15. PMC 4817260. PMID 27034283.
- ↑ Nikoopour, E; Singh, B (2014). "Reciprocity in microbiome and immune system interactions and its implications in disease and health". Inflamm Allergy Drug Targets. 13 (2): 94–104. doi:10.2174/1871528113666140330201056. PMID 24678760.
- ↑ Baird AH, Bhagooli R, Ralph PJ, Takahashi S (2009). "Coral bleaching: the role of the host" (PDF). Trends in Ecology and Evolution. 24 (1): 16–20. doi:10.1016/j.tree.2008.09.005. PMID 19022522.
- ↑ Rosenberg E, Koren O, Reshef L, Efrony R, Zilber-Rosenberg I (2007). "The role of microorganisms in coral health, disease and evolution" (PDF). Nature Reviews Microbiology. 5 (5): 355–362. doi:10.1038/nrmicro1635. PMID 17384666.
- ↑ Leggat W, Ainsworth T, Bythell J, Dove S, Gates R, Hoegh-Guldberg O, Iglesias-Prieto R, Yellowlees D (2007). "The hologenome theory disregards the coral holobiont". Nature Reviews Microbiology. 5 (10): Online Correspondence. doi:10.1038/nrmicro1635-c1.
- ↑ Ainsworth TD, Fine M, Roff G, Hoegh-Guldberg O (2008). "Bacteria are not the primary cause of bleaching in the Mediterranean coral Oculina patagonica". The ISME Journal. 2 (1): 67–73. doi:10.1038/ismej.2007.88. PMID 18059488.
- 1 2 Kuczynski, J.; Lauber, C. L.; Walters, W. A.; Parfrey, L. W.; Clemente, J. C.; Gevers, D.; Knight, R. (2011). "Experimental and analytical tools for studying the human microbiome". Nature Reviews Genetics. 13 (1): 47–58. doi:10.1038/nrg3129. PMID 22179717.
- ↑ Marchesi, J. R. (2010). "Prokaryotic and Eukaryotic Diversity of the Human Gut". Advances in Applied Microbiology Volume 72. Advances in Applied Microbiology. 72. pp. 43–62. doi:10.1016/S0065-2164(10)72002-5. ISBN 9780123809896. PMID 20602987.
- ↑ Vestheim, H.; Jarman, S. N. (2008). "Blocking primers to enhance PCR amplification of rare sequences in mixed samples – a case study on prey DNA in Antarctic krill stomachs". Frontiers in Zoology. 5: 12. doi:10.1186/1742-9994-5-12. PMC 2517594. PMID 18638418.
- ↑ "VAMPS: The Visualization and Analysis of Microbial Population Structures". Bay Paul Center, MBL, Woods Hole. Retrieved 11 March 2012.
- ↑ Caporaso, J. G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F. D.; Costello, E. K.; Fierer, N.; Peña, A. G.; Goodrich, J. K.; Gordon, J. I.; Huttley, G. A.; Kelley, S. T.; Knights, D.; Koenig, J. E.; Ley, R. E.; Lozupone, C. A.; McDonald, D.; Muegge, B. D.; Pirrung, M.; Reeder, J.; Sevinsky, J. R.; Turnbaugh, P. J.; Walters, W. A.; Widmann, J.; Yatsunenko, T.; Zaneveld, J.; Knight, R. (2010). "QIIME allows analysis of high-throughput community sequencing data". Nature Methods. 7 (5): 335–336. doi:10.1038/nmeth.f.303. PMC 3156573. PMID 20383131.
- ↑ Schloss, P. D.; Westcott, S. L.; Ryabin, T.; Hall, J. R.; Hartmann, M.; Hollister, E. B.; Lesniewski, R. A.; Oakley, B. B.; Parks, D. H.; Robinson, C. J.; Sahl, J. W.; Stres, B.; Thallinger, G. G.; Van Horn, D. J.; Weber, C. F. (2009). "Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities". Applied and Environmental Microbiology. 75 (23): 7537–7541. doi:10.1128/AEM.01541-09. PMC 2786419. PMID 19801464.
- ↑ Turnbaugh, P. J.; Hamady, M.; Yatsunenko, T.; Cantarel, B. L.; Duncan, A.; Ley, R. E.; Sogin, M. L.; Jones, W. J.; Roe, B. A.; Affourtit, J. P.; Egholm, M.; Henrissat, B.; Heath, A. C.; Knight, R.; Gordon, J. I. (2008). "A core gut microbiome in obese and lean twins". Nature. 457 (7228): 480–484. Bibcode:2009Natur.457..480T. doi:10.1038/nature07540. PMC 2677729. PMID 19043404.
- ↑ Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K. S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; Mende, D. R.; Li, J.; Xu, J.; Li, S.; Li, D.; Cao, J.; Wang, B.; Liang, H.; Zheng, H.; Xie, Y.; Tap, J.; Lepage, P.; Bertalan, M.; Batto, J. M.; Hansen, T.; Le Paslier, D.; Linneberg, A.; Nielsen, H. B. R.; Pelletier, E.; Renault, P. (2010). "A human gut microbial gene catalogue established by metagenomic sequencing". Nature. 464 (7285): 59–65. Bibcode:2010Natur.464...59.. doi:10.1038/nature08821. PMC 3779803. PMID 20203603.
- ↑ Tringe, S. G.; Von Mering, C.; Kobayashi, A.; Salamov, A. A.; Chen, K.; Chang, H. W.; Podar, M.; Short, J. M.; Mathur, E. J.; Detter, J. C.; Bork, P.; Hugenholtz, P.; Rubin, E. M. (2005). "Comparative Metagenomics of Microbial Communities". Science. 308 (5721): 554–557. Bibcode:2005Sci...308..554T. doi:10.1126/science.1107851. PMID 15845853.
- ↑ Wooley, J. C.; Godzik, A.; Friedberg, I. (2010). Bourne, Philip E., ed. "A Primer on Metagenomics". PLoS Computational Biology. 6 (2): e1000667. Bibcode:2010PLSCB...6E0667W. doi:10.1371/journal.pcbi.1000667. PMC 2829047. PMID 20195499.
- ↑ Muller, J.; Szklarczyk, D.; Julien, P.; Letunic, I.; Roth, A.; Kuhn, M.; Powell, S.; Von Mering, C.; Doerks, T.; Jensen, L. J.; Bork, P. (2009). "EggNOG v2.0: Extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations". Nucleic Acids Research. 38 (Database issue): D190–D195. doi:10.1093/nar/gkp951. PMC 2808932. PMID 19900971.
- ↑ Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. (2009). "KEGG for representation and analysis of molecular networks involving diseases and drugs". Nucleic Acids Research. 38 (Database issue): D355–D360. doi:10.1093/nar/gkp896. PMC 2808910. PMID 19880382.
- ↑ Meyer, F.; Paarmann, D.; d'Souza, M.; Olson, R.; Glass, E. M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; Wilkening, J.; Edwards, R. A. (2008). "The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes". BMC Bioinformatics. 9: 386. doi:10.1186/1471-2105-9-386. PMC 2563014. PMID 18803844.
- ↑ Sun, S.; Chen, J.; Li, W.; Altintas, I.; Lin, A.; Peltier, S.; Stocks, K.; Allen, E. E.; Ellisman, M.; Grethe, J.; Wooley, J. (2010). "Community cyberinfrastructure for Advanced Microbial Ecology Research and Analysis: The CAMERA resource". Nucleic Acids Research. 39 (Database issue): D546–D551. doi:10.1093/nar/gkq1102. PMC 3013694. PMID 21045053.
- ↑ Markowitz, V. M.; Ivanova, N. N.; Szeto, E.; Palaniappan, K.; Chu, K.; Dalevi, D.; Chen, I. M. A.; Grechkin, Y.; Dubchak, I.; Anderson, I.; Lykidis, A.; Mavromatis, K.; Hugenholtz, P.; Kyrpides, N. C. (2007). "IMG/M: A data management and analysis system for metagenomes". Nucleic Acids Research. 36 (Database issue): D534–D538. doi:10.1093/nar/gkm869. PMC 2238950. PMID 17932063.
- ↑ Shi, Y.; Tyson, G. W.; Delong, E. F. (2009). "Metatranscriptomics reveals unique microbial small RNAs in the ocean's water column". Nature. 459 (7244): 266–269. Bibcode:2009Natur.459..266S. doi:10.1038/nature08055. PMID 19444216.
- ↑ Jimenez, R. M.; Delwart, E.; Luptak, A. (2011). "Structure-based Search Reveals Hammerhead Ribozymes in the Human Microbiome". Journal of Biological Chemistry. 286 (10): 7737–7743. doi:10.1074/jbc.C110.209288. PMC 3048661. PMID 21257745.
- ↑ Maron, P. A.; Ranjard, L.; Mougel, C.; Lemanceau, P. (2007). "Metaproteomics: A New Approach for Studying Functional Microbial Ecology". Microbial Ecology. 53 (3): 486–493. doi:10.1007/s00248-006-9196-8. PMID 17431707.
- 1 2 "NIH Human Microbiome Project". US National Institutes of Health, Department of Health and Human Services, US Government. 2016. Retrieved 14 June 2016.
- ↑ Gilbert, J. A.; Meyer, F.; Antonopoulos, D.; Balaji, P.; Brown, C. T.; Brown, C. T.; Desai, N.; Eisen, J. A.; Evers, D.; Field, D.; Feng, W.; Huson, D.; Jansson, J.; Knight, R.; Knight, J.; Kolker, E.; Konstantindis, K.; Kostka, J.; Kyrpides, N.; MacKelprang, R.; McHardy, A.; Quince, C.; Raes, J.; Sczyrba, A.; Shade, A.; Stevens, R. (2010). "Meeting Report: The Terabase Metagenomics Workshop and the Vision of an Earth Microbiome Project". Standards in Genomic Sciences. 3 (3): 243–248. doi:10.4056/sigs.1433550. PMC 3035311. PMID 21304727.
- ↑ Gilbert, J. A.; O'Dor, R.; King, N.; Vogel, T. M. (2011). "The importance of metagenomic surveys to microbial ecology: Or why Darwin would have been a metagenomic scientist". Microbial Informatics and Experimentation. 1 (1): 5. doi:10.1186/2042-5783-1-5. PMC 3348666. PMID 22587826.
- ↑ magazine, Ewen. "Microbial DNA in Human Body Can Be Used to Identify Individuals". Retrieved 2015-05-17.
- ↑ "Microbiomes raise privacy concerns". Retrieved 2015-05-17.
- ↑ Franzosa, Eric A.; Huang, Katherine; Meadow, James F.; Gevers, Dirk; Lemon, Katherine P.; Bohannan, Brendan J. M.; Huttenhower, Curtis (2015-05-11). "Identifying personal microbiomes using metagenomic codes". Proceedings of the National Academy of Sciences. 112 (22): E2930–8. Bibcode:2015PNAS..112E2930F. doi:10.1073/pnas.1423854112. ISSN 0027-8424. PMC 4460507. PMID 25964341.
- ↑ Yong, Ed. "Can The Microbes You Leave Behind Be Used to Identify You?". Retrieved 2015-05-17.
External links
| Look up microbiota in Wiktionary, the free dictionary. |