Proteasome

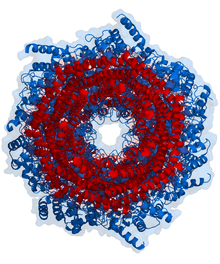
Proteasomes are protein complexes inside all eukaryotes and archaea, and in some bacteria. In eukaryotes, proteasomes are located in the nucleus and the cytoplasm.[1] The main function of the proteasome is to degrade unneeded or damaged proteins by proteolysis, a chemical reaction that breaks peptide bonds. Enzymes that help such reactions are called proteases. Proteasomes are part of a major mechanism by which cells regulate the concentration of particular proteins and degrade misfolded proteins. The degradation process yields peptides of about seven to eight amino acids long, which can then be further degraded into shorter amino acid sequences and used in synthesizing new proteins.[2] Proteins are tagged for degradation with a small protein called ubiquitin. The tagging reaction is catalyzed by enzymes called ubiquitin ligases. Once a protein is tagged with a single ubiquitin molecule, this is a signal to other ligases to attach additional ubiquitin molecules. The result is a polyubiquitin chain that is bound by the proteasome, allowing it to degrade the tagged protein.[2]
In structure, the proteasome is a cylindrical complex containing a "core" of four stacked rings forming a central pore. Each ring is composed of seven individual proteins. The inner two rings are made of seven β subunits that contain three to seven protease active sites. These sites are located on the interior surface of the rings, so that the target protein must enter the central pore before it is degraded. The outer two rings each contain seven α subunits whose function is to maintain a "gate" through which proteins enter the barrel. These α subunits are controlled by binding to "cap" structures or regulatory particles that recognize polyubiquitin tags attached to protein substrates and initiate the degradation process. The overall system of ubiquitination and proteasomal degradation is known as the ubiquitin-proteasome system.[3]
The proteasomal degradation pathway is essential for many cellular processes, including the cell cycle, the regulation of gene expression, and responses to oxidative stress. The importance of proteolytic degradation inside cells and the role of ubiquitin in proteolytic pathways was acknowledged in the award of the 2004 Nobel Prize in Chemistry to Aaron Ciechanover, Avram Hershko and Irwin Rose.[4]
Discovery
Before the discovery of the ubiquitin proteasome system, protein degradation in cells was thought to rely mainly on lysosomes, membrane-bound organelles with acidic and protease-filled interiors that can degrade and then recycle exogenous proteins and aged or damaged organelles.[2] However, work by Alfred Goldberg in 1977 on ATP-dependent protein degradation in reticulocytes, which lack lysosomes, suggested the presence of a second intracellular degradation mechanism.[5] This was shown in 1978 to be composed of several distinct protein chains, a novelty among proteases at the time.[6] Later work on modification of histones led to the identification of an unexpected covalent modification of the histone protein by a bond between a lysine side chain of the histone and the C-terminal glycine residue of ubiquitin, a protein that had no known function.[7] It was then discovered that a previously identified protein associated with proteolytic degradation, known as ATP-dependent proteolysis factor 1 (APF-1), was the same protein as ubiquitin.[8] The proteolytic activities of this system was isolated as a multi-protein complex originally called the multi-catalytic proteinase complex by Sherwin Wilk and Marion Orlowski.[9] Later, the ATP-dependent proteolytic complex that was responsible for ubiquitin-dependent protein degradation was discovered and was called the 26S proteasome.[10][11]
Much of the early work leading up to the discovery of the ubiquitin proteasome system occurred in the late 1970s and early 1980s at the Technion in the laboratory of Avram Hershko, where Aaron Ciechanover worked as a graduate student. Hershko's year-long sabbatical in the laboratory of Irwin Rose at the Fox Chase Cancer Center provided key conceptual insights, though Rose later downplayed his role in the discovery.[12] The three shared the 2004 Nobel Prize in Chemistry for their work in discovering this system.[4]
Although electron microscopy data revealing the stacked-ring structure of the proteasome became available in the mid-1980s,[13] the first structure of the proteasome core particle was not solved by X-ray crystallography until 1994.[14]
Structure and organization
The proteasome subcomponents are often referred to by their Svedberg sedimentation coefficient (denoted S). The proteasome most exclusively used in mammals is the cytosolic 26S proteasome, which is about 2000 kilodaltons (kDa) in molecular mass containing one 20S protein subunit and two 19S regulatory cap subunits. The core is hollow and provides an enclosed cavity in which proteins are degraded; openings at the two ends of the core allow the target protein to enter. Each end of the core particle associates with a 19S regulatory subunit that contains multiple ATPase active sites and ubiquitin binding sites; it is this structure that recognizes polyubiquitinated proteins and transfers them to the catalytic core. An alternative form of regulatory subunit called the 11S particle can associate with the core in essentially the same manner as the 19S particle; the 11S may play a role in degradation of foreign peptides such as those produced after infection by a virus.[15]
20S core particle
The number and diversity of subunits contained in the 20S core particle depends on the organism; the number of distinct and specialized subunits is larger in multicellular than unicellular organisms and larger in eukaryotes than in prokaryotes. All 20S particles consist of four stacked heptameric ring structures that are themselves composed of two different types of subunits; α subunits are structural in nature, whereas β subunits are predominantly catalytic. The outer two rings in the stack consist of seven α subunits each, which serve as docking domains for the regulatory particles and the alpha subunits N-termini form a gate that blocks unregulated access of substrates to the interior cavity.[16] The inner two rings each consist of seven β subunits and contain the protease active sites that perform the proteolysis reactions. Three distinct catalytic activities were identified in the purified complex: chymotrypsin-like, trypsin-like and peptidylglutamyl-peptide hydrolyzing.[17] The size of the proteasome is relatively conserved and is about 150 angstroms (Å) by 115 Å. The interior chamber is at most 53 Å wide, though the entrance can be as narrow as 13 Å, suggesting that substrate proteins must be at least partially unfolded to enter.[18]
In archaea such as Thermoplasma acidophilum, all the α and all the β subunits are identical, whereas eukaryotic proteasomes such as those in yeast contain seven distinct types of each subunit. In mammals, the β1, β2, and β5 subunits are catalytic; although they share a common mechanism, they have three distinct substrate specificities considered chymotrypsin-like, trypsin-like, and peptidyl-glutamyl peptide-hydrolyzing (PHGH).[19] Alternative β forms denoted β1i, β2i, and β5i can be expressed in hematopoietic cells in response to exposure to pro-inflammatory signals such as cytokines, in particular, interferon gamma. The proteasome assembled with these alternative subunits is known as the immunoproteasome, whose substrate specificity is altered relative to the normal proteasome.[18] Recently an alternative proteasome was identified in human cells that lack the α3 core subunit.[20] These proteasomes (known as the α4-α4 proteasomes) instead form 20S core particles containing an additional α4 subunit in place of the missing α3 subunit. Interestingly, these alternative 'α4-α4' proteasomes have been known previously to exist in yeast.[21] Although the precise function of these proteasome isoforms is still largely unknown, cells expressing these proteasomes show enhanced resistance to toxicity induced by metallic ions such as cadmium.[20][22]
19S regulatory particle
The 19S particle in eukaryotes consists of 19 individual proteins and is divisible into two subassemblies, a 9-subunit base that binds directly to the α ring of the 20S core particle, and a 10-subunit lid. Six of the nine base proteins are ATPase subunits from the AAA Family, and an evolutionary homolog of these ATPases exists in archaea, called PAN (Proteasome-Activating Nucleotidase).[23] The association of the 19S and 20S particles requires the binding of ATP to the 19S ATPase subunits, and ATP hydrolysis is required for the assembled complex to degrade folded and ubiquitinated proteins. Note that only the step of substrate unfolding requires energy from ATP hydrolysis, while ATP-binding alone can support all the other steps required for protein degradation (e.g., complex assembly, gate opening, translocation, and proteolysis).[24][25] In fact, ATP binding to the ATPases by itself supports the rapid degradation of unfolded proteins. However, while ATP hydrolysis is required for unfolding only, it is not yet clear whether this energy may be used in the coupling of some of these steps.[25][26]
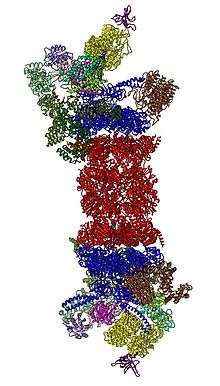
In 2012, two independent efforts have elucidated the molecular architecture of the 26S proteasome by single particle electron microscopy.[28][29] More recently, a pseudo-atomic atomic model has been built, again using cryo-EM.[27] In the heart of the 19S, directly adjacent to the 20S, are the AAA-ATPases (AAA proteins) that assemble to a heterohexameric ring of the order Rpt1/Rpt2/Rpt6/Rpt3/Rpt4/Rpt5. This ring is a trimer of dimers: Rpt1/Rpt2, Rpt6/Rpt3, and Rpt4/Rpt5 dimerize via their N-terminal coiled-coils. These coiled-coils protrude from the hexameric ring. The largest regulatory particle non-ATPases Rpn1 and Rpn2 bind to the tips of Rpt1/2 and Rpt6/3, respectively. The ubiquitin receptor Rpn13 binds to Rpn2 and completes the base cub-complex. The lid covers one half of the AAA-ATPase hexamer (Rpt6/Rpt3/Rpt4) and, unexpectedly, directly contacts the 20S via Rpn6 and to lesser extent Rpn5. The subunits Rpn9, Rpn5, Rpn6, Rpn7, Rpn3, and Rpn12, which are structurally related among themselves and to subunits of the COP9 complex and the Eukaryotic initiation factor 3 (hence called PCI subunits) assemble to a horseshoe-like structure enclosing the Rpn8/Rpn11 heterodimer. Rpn11, the deubiquinating enzyme, is placed at the mouth of the AAA-ATPase hexamer, ideally positioned to remove ubiquitin moieties immediately before translocation of substrates into the 20S. The second ubiquitin receptor identified to date, Rpn10, is positioned at the periphery of the lid, near subunits Rpn8 and Rpn9.
Conformational changes of 19S
The 19S regulatory particle has been observed in three strongly differing conformational states to date.[30] Realization of all these three conformational states is likely necessary for accomplishing substrate recognition and degradation (see below). A hallmark of the AAA-ATPase configuration in this predominant low-energy state is a staircase- or lockwasher-like arrangement of the AAA-domains.[27][28] Also in the presence of ATP but absence of substrate an alternative, less abundant conformation of the 19S is adopted primarily differing in the positioning of the lid with respect to the AAA-ATPase module.[30] In the presence of ATP-gammaS or a substrate (stabilized in a 26S mutant with defective Rpn11) a third conformation has been observed displaying a dramatic structural change of the AAA-ATPase module.[31][32]
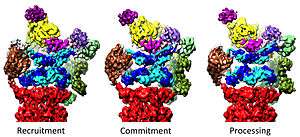
Regulation of the 20S by the 19S
The 19S regulatory particle is responsible for stimulating the 20S to degrade proteins. A primary function of the 19S regulatory ATPases is to open the gate in the 20S that blocks the entry of substrates into the degradation chamber.[33] The mechanism by which the proteasomal ATPase open this gate has been recently elucidated.[16] 20S gate opening, and thus substrate degradation, requires the C-termini of the proteasomal ATPases, which contains a specific motif (i.e., HbYX motif). The ATPases C-termini bind into pockets in the top of the 20S, and tether the ATPase complex to the 20S proteolytic complex, thus joining the substrate unfolding equipment with the 20S degradation machinery. Binding of these C-termini into these 20S pockets by themselves stimulates opening of the gate in the 20S in much the same way that a "key-in-a-lock" opens a door.[16] The precise mechanism by which this "key-in-a-lock" mechanism functions has been structurally elucidated.[34]
11S regulatory particle
20S proteasomes can also associate with a second type of regulatory particle, the 11S regulatory particle, a heptameric structure that does not contain any ATPases and can promote the degradation of short peptides but not of complete proteins. It is presumed that this is because the complex cannot unfold larger substrates. This structure is also known as PA28 or REG. The mechanisms by which it binds to the core particle through the C-terminal tails of its subunits and induces α-ring conformational changes to open the 20S gate suggest a similar mechanism for the 19S particle.[35] The expression of the 11S particle is induced by interferon gamma and is responsible, in conjunction with the immunoproteasome β subunits, for the generation of peptides that bind to the major histocompatibility complex.[15]
Assembly
The assembly of the proteasome is a complex process due to the number of subunits that must associate to form an active complex. The β subunits are synthesized with N-terminal "propeptides" that are post-translationally modified during the assembly of the 20S particle to expose the proteolytic active site. The 20S particle is assembled from two half-proteasomes, each of which consists of a seven-membered pro-β ring attached to a seven-membered α ring. The association of the β rings of the two half-proteasomes triggers threonine-dependent autolysis of the propeptides to expose the active site. These β interactions are mediated mainly by salt bridges and hydrophobic interactions between conserved alpha helices whose disruption by mutation damages the proteasome's ability to assemble.[36] The assembly of the half-proteasomes, in turn, is initiated by the assembly of the α subunits into their heptameric ring, forming a template for the association of the corresponding pro-β ring. The assembly of α subunits has not been characterized.[37]
Only recently, the assembly process of the 19S regulatory particle has been elucidated to considerable extent. The 19S regulatory particle assembles as two distinct subcomponents, the base and the lid. Assembly of the base complex is facilitated by four assembly chaperones, Hsm3/S5b, Nas2/p27, Rpn14/PAAF1, and Nas6/gankyrin (names for yeast/mammals).[38] These assembly chaperones bind to the AAA-ATPase subunits and their main function seems to be to ensure proper assembly of the heterohexameric AAA-ATPase ring. To date it is still under debate whether the base complex assembles separately, whether the assembly is templated by the 20S core particle, or whether alternative assembly pathways exist. In addition to the four assembly chaperones, the deubiquitinating enzyme Ubp6/Usp14 also promotes base assembly, but it is not essential.[39] The lid assembles separately in a specific order and does not require assembly chaperones.[40]
The protein degradation process
Ubiquitination and targeting
Proteins are targeted for degradation by the proteasome with covalent modification of a lysine residue that requires the coordinated reactions of three enzymes. In the first step, a ubiquitin-activating enzyme (known as E1) hydrolyzes ATP and adenylylates a ubiquitin molecule. This is then transferred to E1's active-site cysteine residue in concert with the adenylylation of a second ubiquitin.[41] This adenylylated ubiquitin is then transferred to a cysteine of a second enzyme, ubiquitin-conjugating enzyme (E2). In the last step, a member of a highly diverse class of enzymes known as ubiquitin ligases (E3) recognizes the specific protein to be ubiquitinated and catalyzes the transfer of ubiquitin from E2 to this target protein. A target protein must be labeled with at least four ubiquitin monomers (in the form of a polyubiquitin chain) before it is recognized by the proteasome lid.[42] It is therefore the E3 that confers substrate specificity to this system.[43] The number of E1, E2, and E3 proteins expressed depends on the organism and cell type, but there are many different E3 enzymes present in humans, indicating that there is a huge number of targets for the ubiquitin proteasome system.
The mechanism by which a polyubiquitinated protein is targeted to the proteasome is not fully understood. Ubiquitin-receptor proteins have an N-terminal ubiquitin-like (UBL) domain and one or more ubiquitin-associated (UBA) domains. The UBL domains are recognized by the 19S proteasome caps and the UBA domains bind ubiquitin via three-helix bundles. These receptor proteins may escort polyubiquitinated proteins to the proteasome, though the specifics of this interaction and its regulation are unclear.[44]
The ubiquitin protein itself is 76 amino acids long and was named due to its ubiquitous nature, as it has a highly conserved sequence and is found in all known eukaryotic organisms.[45] The genes encoding ubiquitin in eukaryotes are arranged in tandem repeats, possibly due to the heavy transcription demands on these genes to produce enough ubiquitin for the cell. It has been proposed that ubiquitin is the slowest-evolving protein identified to date.[46] Ubiquitin contains seven lysine residues to which another ubiquitin can be ligated, resulting in different types of polyubiquitin chains.[47] Chains in which each additional ubiquitin is linked to lysine 48 of the previous ubiquitin have a role in proteasome targeting, while other types of chains may be involved in other processes.[48][49]
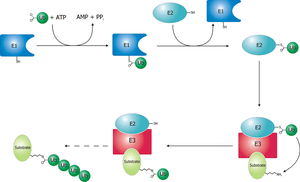
Unfolding and translocation
After a protein has been ubiquitinated, it is recognized by the 19S regulatory particle in an ATP-dependent binding step.[25] The substrate protein must then enter the interior of the 20S particle to come in contact with the proteolytic active sites. Because the 20S particle's central channel is narrow and gated by the N-terminal tails of the α ring subunits, the substrates must be at least partially unfolded before they enter the core. The passage of the unfolded substrate into the core is called translocation and necessarily occurs after deubiquitination.[25] However, the order in which substrates are deubiquitinated and unfolded is not yet clear.[50] Which of these processes is the rate-limiting step in the overall proteolysis reaction depends on the specific substrate; for some proteins, the unfolding process is rate-limiting, while deubiquitination is the slowest step for other proteins.[24] The extent to which substrates must be unfolded before translocation is not known, but substantial tertiary structure, and in particular nonlocal interactions such as disulfide bonds, are sufficient to inhibit degradation.[51] The presence of intrinsically disordered protein segments of sufficient size, either at the protein terminus or internally, has also been proposed to facilitate efficient initiation of degradation.[52][53]
The gate formed by the α subunits prevents peptides longer than about four residues from entering the interior of the 20S particle. The ATP molecules bound before the initial recognition step are hydrolyzed before translocation. While energy is needed for substrate unfolding, it is not required for translocation.[24][25] The assembled 26S proteasome can degrade unfolded proteins in the presence of a non-hydrolyzable ATP analog, but cannot degrade folded proteins, indicating that energy from ATP hydrolysis is used for substrate unfolding.[24] Passage of the unfolded substrate through the opened gate occurs via facilitated diffusion if the 19S cap is in the ATP-bound state.[54]
The mechanism for unfolding of globular proteins is necessarily general, but somewhat dependent on the amino acid sequence. Long sequences of alternating glycine and alanine have been shown to inhibit substrate unfolding, decreasing the efficiency of proteasomal degradation; this results in the release of partially degraded byproducts, possibly due to the decoupling of the ATP hydrolysis and unfolding steps.[55] Such glycine-alanine repeats are also found in nature, for example in silk fibroin; in particular, certain Epstein–Barr virus gene products bearing this sequence can stall the proteasome, helping the virus propagate by preventing antigen presentation on the major histocompatibility complex.[56]

Proteolysis
The mechanism of proteolysis by the β subunits of the 20S core particle is through a threonine-dependent nucleophilic attack. This mechanism may depend on an associated water molecule for deprotonation of the reactive threonine hydroxyl. Degradation occurs within the central chamber formed by the association of the two β rings and normally does not release partially degraded products, instead reducing the substrate to short polypeptides typically 7–9 residues long, though they can range from 4 to 25 residues, depending on the organism and substrate. The biochemical mechanism that determines product length is not fully characterized.[57] Although the three catalytic β subunits have a common mechanism, they have slightly different substrate specificities, which are considered chymotrypsin-like, trypsin-like, and peptidyl-glutamyl peptide-hydrolyzing (PHGH)-like. These variations in specificity are the result of interatomic contacts with local residues near the active sites of each subunit. Each catalytic β subunit also possesses a conserved lysine residue required for proteolysis.[19]
Although the proteasome normally produces very short peptide fragments, in some cases these products are themselves biologically active and functional molecules. Certain transcription factors regulating the expression of specific genes, including one component of the mammalian complex NF-κB, are synthesized as inactive precursors whose ubiquitination and subsequent proteasomal degradation converts them to an active form. Such activity requires the proteasome to cleave the substrate protein internally, rather than processively degrading it from one terminus. It has been suggested that long loops on these proteins' surfaces serve as the proteasomal substrates and enter the central cavity, while the majority of the protein remains outside.[58] Similar effects have been observed in yeast proteins; this mechanism of selective degradation is known as regulated ubiquitin/proteasome dependent processing (RUP).[59]
Ubiquitin-independent degradation
Although most proteasomal substrates must be ubiquitinated before being degraded, there are some exceptions to this general rule, especially when the proteasome plays a normal role in the post-translational processing of the protein. The proteasomal activation of NF-κB by processing p105 into p50 via internal proteolysis is one major example.[58] Some proteins that are hypothesized to be unstable due to intrinsically unstructured regions,[60] are degraded in a ubiquitin-independent manner. The most well-known example of a ubiquitin-independent proteasome substrate is the enzyme ornithine decarboxylase.[61] Ubiquitin-independent mechanisms targeting key cell cycle regulators such as p53 have also been reported, although p53 is also subject to ubiquitin-dependent degradation.[62] Finally, structurally abnormal, misfolded, or highly oxidized proteins are also subject to ubiquitin-independent and 19S-independent degradation under conditions of cellular stress.[63]
Evolution

The 20S proteasome is both ubiquitous and essential in eukaryotes. Some prokaryotes, including many archaea and the bacterial order Actinomycetales also share homologs of the 20S proteasome, whereas most bacteria possess heat shock genes hslV and hslU, whose gene products are a multimeric protease arranged in a two-layered ring and an ATPase.[64] The hslV protein has been hypothesized to resemble the likely ancestor of the 20S proteasome.[65] In general, HslV is not essential in bacteria, and not all bacteria possess it, whereas some protists possess both the 20S and the hslV systems.[64] Many bacteria also possess other homologs of the proteasome and an associated ATPase, most notably ClpP and ClpX. This redundancy explains why the HslUV system is not essential.
Sequence analysis suggests that the catalytic β subunits diverged earlier in evolution than the predominantly structural α subunits. In bacteria that express a 20S proteasome, the β subunits have high sequence identity to archaeal and eukaryotic β subunits, whereas the α sequence identity is much lower. The presence of 20S proteasomes in bacteria may result from lateral gene transfer, while the diversification of subunits among eukaryotes is ascribed to multiple gene duplication events.[64]
Cell cycle control
Cell cycle progression is controlled by ordered action of cyclin-dependent kinases (CDKs), activated by specific cyclins that demarcate phases of the cell cycle. Mitotic cyclins, which persist in the cell for only a few minutes, have one of the shortest life spans of all intracellular proteins.[2] After a CDK-cyclin complex has performed its function, the associated cyclin is polyubiquitinated and destroyed by the proteasome, which provides directionality for the cell cycle. In particular, exit from mitosis requires the proteasome-dependent dissociation of the regulatory component cyclin B from the mitosis promoting factor complex.[66] In vertebrate cells, "slippage" through the mitotic checkpoint leading to premature M phase exit can occur despite the delay of this exit by the spindle checkpoint.[67]
Earlier cell cycle checkpoints such as post-restriction point check between G1 phase and S phase similarly involve proteasomal degradation of cyclin A, whose ubiquitination is promoted by the anaphase promoting complex (APC), an E3 ubiquitin ligase.[68] The APC and the Skp1/Cul1/F-box protein complex (SCF complex) are the two key regulators of cyclin degradation and checkpoint control; the SCF itself is regulated by the APC via ubiquitination of the adaptor protein, Skp2, which prevents SCF activity before the G1-S transition.[69]
Individual components of the 19S particle have their own regulatory roles. Gankyrin, a recently identified oncoprotein, is one of the 19S subcomponents that also tightly binds the cyclin-dependent kinase CDK4 and plays a key role in recognizing ubiquitinated p53, via its affinity for the ubiquitin ligase MDM2. Gankyrin is anti-apoptotic and has been shown to be overexpressed in some tumor cell types such as hepatocellular carcinoma.[70]
Regulation of plant growth
In plants, signaling by auxins, or phytohormones that order the direction and tropism of plant growth, induces the targeting of a class of transcription factor repressors known as Aux/IAA proteins for proteasomal degradation. These proteins are ubiquitinated by SCFTIR1, or SCF in complex with the auxin receptor TIR1. Degradation of Aux/IAA proteins derepresses transcription factors in the auxin-response factor (ARF) family and induces ARF-directed gene expression.[71] The cellular consequences of ARF activation depend on the plant type and developmental stage, but are involved in directing growth in roots and leaf veins. The specific response to ARF derepression is thought to be mediated by specificity in the pairing of individual ARF and Aux/IAA proteins.[72]
Apoptosis
Both internal and external signals can lead to the induction of apoptosis, or programmed cell death. The resulting deconstruction of cellular components is primarily carried out by specialized proteases known as caspases, but the proteasome also plays important and diverse roles in the apoptotic process. The involvement of the proteasome in this process is indicated by both the increase in protein ubiquitination, and of E1, E2, and E3 enzymes that is observed well in advance of apoptosis.[73][74][75] During apoptosis, proteasomes localized to the nucleus have also been observed to translocate to outer membrane blebs characteristic of apoptosis.[76]
Proteasome inhibition has different effects on apoptosis induction in different cell types. In general, the proteasome is not required for apoptosis, although inhibiting it is pro-apoptotic in most cell types that have been studied. Apoptosis is mediated through disrupting the regulated degradation of pro-growth cell cycle proteins.[77] However, some cell lines — in particular, primary cultures of quiescent and differentiated cells such as thymocytes and neurons — are prevented from undergoing apoptosis on exposure to proteasome inhibitors. The mechanism for this effect is not clear, but is hypothesized to be specific to cells in quiescent states, or to result from the differential activity of the pro-apoptotic kinase JNK.[78] The ability of proteasome inhibitors to induce apoptosis in rapidly dividing cells has been exploited in several recently developed chemotherapy agents such as bortezomib and salinosporamide A.
Response to cellular stress
In response to cellular stresses – such as infection, heat shock, or oxidative damage – heat shock proteins that identify misfolded or unfolded proteins and target them for proteasomal degradation are expressed. Both Hsp27 and Hsp90—chaperone proteins have been implicated in increasing the activity of the ubiquitin-proteasome system, though they are not direct participants in the process.[79] Hsp70, on the other hand, binds exposed hydrophobic patches on the surface of misfolded proteins and recruits E3 ubiquitin ligases such as CHIP to tag the proteins for proteasomal degradation.[80] The CHIP protein (carboxyl terminus of Hsp70-interacting protein) is itself regulated via inhibition of interactions between the E3 enzyme CHIP and its E2 binding partner.[81]
Similar mechanisms exist to promote the degradation of oxidatively damaged proteins via the proteasome system. In particular, proteasomes localized to the nucleus are regulated by PARP and actively degrade inappropriately oxidized histones.[82] Oxidized proteins, which often form large amorphous aggregates in the cell, can be degraded directly by the 20S core particle without the 19S regulatory cap and do not require ATP hydrolysis or tagging with ubiquitin.[63] However, high levels of oxidative damage increases the degree of cross-linking between protein fragments, rendering the aggregates resistant to proteolysis. Larger numbers and sizes of such highly oxidized aggregates are associated with aging.[83]
Dysregulation of the ubiquitin proteasome system may contribute to several neural diseases. It may lead to brain tumors such as astrocytomas.[84] In some of the late-onset neurodegenerative diseases that share aggregation of misfolded proteins as a common feature, such as Parkinson's disease and Alzheimer's disease, large insoluble aggregates of misfolded proteins can form and then result in neurotoxicity, through mechanisms that are not yet well understood. Decreased proteasome activity has been suggested as a cause of aggregation and Lewy body formation in Parkinson's.[85] This hypothesis is supported by the observation that yeast models of Parkinson's are more susceptible to toxicity from α-synuclein, the major protein component of Lewy bodies, under conditions of low proteasome activity.[86] Impaired proteasomal activity may underlie cognitive disorders such as the autism spectrum disorders, and muscle and nerve diseases such as inclusion body myopathy.[84]
Role in the immune system
The proteasome plays a straightforward but critical role in the function of the adaptive immune system. Peptide antigens are displayed by the major histocompatibility complex class I (MHC) proteins on the surface of antigen-presenting cells. These peptides are products of proteasomal degradation of proteins originated by the invading pathogen. Although constitutively expressed proteasomes can participate in this process, a specialized complex composed of proteins, whose expression is induced by interferon gamma, are the primary producers of peptides which are optimal in size and composition for MHC binding. These proteins whose expression increases during the immune response include the 11S regulatory particle, whose main known biological role is regulating the production of MHC ligands, and specialized β subunits called β1i, β2i, and β5i with altered substrate specificity. The complex formed with the specialized β subunits is known as the immunoproteasome.[15] Another β5i variant subunit, β5t, is expressed in the thymus, leading to a thymus-specific "thymoproteasome" whose function is as yet unclear.[87]
The strength of MHC class I ligand binding is dependent on the composition of the ligand C-terminus, as peptides bind by hydrogen bonding and by close contacts with a region called the "B pocket" on the MHC surface. Many MHC class I alleles prefer hydrophobic C-terminal residues, and the immunoproteasome complex is more likely to generate hydrophobic C-termini.[88]
Due to its role in generating the activated form of NF-κB, an anti-apoptotic and pro-inflammatory regulator of cytokine expression, proteasomal activity has been linked to inflammatory and autoimmune diseases. Increased levels of proteasome activity correlate with disease activity and have been implicated in autoimmune diseases including systemic lupus erythematosus and rheumatoid arthritis.[15]
The proteasome is also involved in Intracellular antibody-mediated proteolysis of antibody-bound virions. In this neutralisation pathway, TRIM21 (a protein of the tripartite motif family) binds with immunoglobulin G to direct the virion to the proteasome where it is degraded.[89]
Proteasome inhibitors
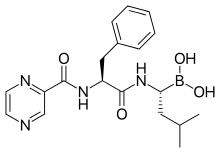

Proteasome inhibitors have effective anti-tumor activity in cell culture, inducing apoptosis by disrupting the regulated degradation of pro-growth cell cycle proteins.[77] This approach of selectively inducing apoptosis in tumor cells has proven effective in animal models and human trials.
Lactacystin, a natural product synthesized by Streptomyces bacteria, was the first non-peptidic proteasome inhibitor discovered[90] and is widely used as a research tool in biochemistry and cell biology. Lactacystin was licensed to Myogenics/Proscript, which was acquired by Millennium Pharmaceuticals, now part of Takeda Pharmaceuticals. Lactacystin covalently modifies the amino-terminal threonine of catalytic β subunits of the proteasome, particularly the β5 subunit responsible for the proteasome's chymotrypsin-like activity. This discovery helped to establish the proteasome as a mechanistically novel class of protease: an amino-terminal threonine protease.
Bortezomib (Boronated MG132), a molecule developed by Millennium Pharmaceuticals and marketed as Velcade, is the first proteasome inhibitor to reach clinical use as a chemotherapy agent.[91] Bortezomib is used in the treatment of multiple myeloma.[92] Notably, multiple myeloma has been observed to result in increased proteasome-derived peptide levels in blood serum that decrease to normal levels in response to successful chemotherapy.[93] Studies in animals have indicated that bortezomib may also have clinically significant effects in pancreatic cancer.[94][95] Preclinical and early clinical studies have been started to examine bortezomib's effectiveness in treating other B-cell-related cancers,[96] particularly some types of non-Hodgkin's lymphoma.[97] Clinical results also seem to justify use of proteasome inhibitor combined with chemotherapy, for B-cell acute lymphoblastic leukemia [98] Proteasome inhibitor can kill some types of cultured leukemia cells that are resistant to glucocorticoid.[99]
The molecule ritonavir, marketed as Norvir, was developed as a protease inhibitor and used to target HIV infection. However, it has been shown to inhibit proteasomes as well as free proteases; to be specific, the chymotrypsin-like activity of the proteasome is inhibited by ritonavir, while the trypsin-like activity is somewhat enhanced.[100] Studies in animal models suggest that ritonavir may have inhibitory effects on the growth of glioma cells.[101]
Proteasome inhibitors have also shown promise in treating autoimmune diseases in animal models. For example, studies in mice bearing human skin grafts found a reduction in the size of lesions from psoriasis after treatment with a proteasome inhibitor.[102] Inhibitors also show positive effects in rodent models of asthma.[103]
Labeling and inhibition of the proteasome is also of interest in laboratory settings for both in vitro and in vivo study of proteasomal activity in cells. The most commonly used laboratory inhibitors are lactacystin and the peptide aldehyde MG132 initially developed by Goldberg lab. Fluorescent inhibitors have also been developed to specifically label the active sites of the assembled proteasome.[104]
Clinical significance
The Proteasome and its subunits are of clinical significance for at least two reasons: (1) a compromised complex assembly or a dysfunctional proteasome can be associated with the underlying pathophysiology of specific diseases, and (2) they can be exploited as drug targets for therapeutic interventions. More recently, more effort has been made to consider the proteasome for the development of novel diagnostic markers and strategies. An improved and comprehensive understanding of the pathophysiology of the proteasome should lead to clinical applications in the future.
The proteasomes form a pivotal component for the Ubiquitin-Proteasome System (UPS) [105] and corresponding cellular Protein Quality Control (PQC). Protein ubiquitination and subsequent proteolysis and degradation by the proteasome are important mechanisms in the regulation of the cell cycle, cell growth and differentiation, gene transcription, signal transduction and apoptosis.[106] Subsequently, a compromised proteasome complex assembly and function lead to reduced proteolytic activities and the accumulation of damaged or misfolded protein species. Such protein accumulation may contribute to the pathogenesis and phenotypic characteristics in neurodegenerative diseases,[107][108] cardiovascular diseases,[109][110][111] inflammatory responses and autoimmune diseases,[112] and systemic DNA damage responses leading to malignancies.[113]
Several experimental and clinical studies have indicated that aberrations and deregulations of the UPS contribute to the pathogenesis of several neurodegenerative and myodegenerative disorders, including Alzheimer's disease,[114] Parkinson's disease[115] and Pick's disease,[116] Amyotrophic lateral sclerosis (ALS),[116] Huntington's disease,[115] Creutzfeldt–Jakob disease,[117] and motor neuron diseases, polyglutamine (PolyQ) diseases, Muscular dystrophies[118] and several rare forms of neurodegenerative diseases associated with dementia.[119] As part of the Ubiquitin-Proteasome System (UPS), the proteasome maintains cardiac protein homeostasis and thus plays a significant role in cardiac Ischemic injury,[120] ventricular hypertrophy[121] and Heart failure.[122] Additionally, evidence is accumulating that the UPS plays an essential role in malignant transformation. UPS proteolysis plays a major role in responses of cancer cells to stimulatory signals that are critical for the development of cancer. Accordingly, gene expression by degradation of transcription factors, such as p53, c-Jun, c-Fos, NF-κB, c-Myc, HIF-1α, MATα2, STAT3, sterol-regulated element-binding proteins and androgen receptors are all controlled by the UPS and thus involved in the development of various malignancies.[123] Moreover, the UPS regulates the degradation of tumor suppressor gene products such as adenomatous polyposis coli (APC) in colorectal cancer, retinoblastoma (Rb). and von Hippel-Lindau tumor suppressor (VHL), as well as a number of proto-oncogenes (Raf, Myc, Myb, Rel, Src, Mos, Abl). The UPS is also involved in the regulation of inflammatory responses. This activity is usually attributed to the role of proteasomes in the activation of NF-κB which further regulates the expression of pro inflammatory cytokines such as TNF-α, IL-β, IL-8, adhesion molecules (ICAM-1, VCAM-1, P selectine) and prostaglandins and nitric oxide (NO).[112] Additionally, the UPS also plays a role in inflammatory responses as regulators of leukocyte proliferation, mainly through proteolysis of cyclines and the degradation of CDK inhibitors.[124] Lastly, autoimmune disease patients with SLE, Sjogren's syndrome and rheumatoid arthritis (RA) predominantly exhibit circulating proteasomes which can be applied as clinical biomarkers.[125]
See also
References
- ↑ Peters JM, Franke WW, Kleinschmidt JA (Mar 1994). "Distinct 19 S and 20 S subcomplexes of the 26 S proteasome and their distribution in the nucleus and the cytoplasm". The Journal of Biological Chemistry. 269 (10): 7709–18. PMID 8125997.
- 1 2 3 4 Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J (2004). "3". Molecular cell biology (5th ed.). New York: W.H. Freeman and CO. pp. 66–72. ISBN 978-0-7167-4366-8.
- ↑ Nassif, Nicholas D.; Cambray, Samantha E.; Kraut, Daniel A. (May 2014). "Slipping up: Partial substrate degradation by ATP-dependent proteases". IUBMB Life. 66 (5): 309–317. doi:10.1002/iub.1271. Retrieved 15 April 2016.
- 1 2 Nobel Prize Committee (2004). "Nobel Prize Awardees in Chemistry, 2004". Retrieved 2006-12-11.
- ↑ Etlinger JD, Goldberg AL (Jan 1977). "A soluble ATP-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes". Proceedings of the National Academy of Sciences of the United States of America. 74 (1): 54–8. doi:10.1073/pnas.74.1.54. PMC 393195
 . PMID 264694.
. PMID 264694. - ↑ Ciehanover A, Hod Y, Hershko A (Apr 1978). "A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes". Biochemical and Biophysical Research Communications. 81 (4): 1100–5. doi:10.1016/0006-291X(78)91249-4. PMID 666810.
- ↑ Goldknopf IL, Busch H (Mar 1977). "Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate-protein A24". Proceedings of the National Academy of Sciences of the United States of America. 74 (3): 864–8. doi:10.1073/pnas.74.3.864. PMC 430507. PMID 265581.
- ↑ Ciechanover A (Sep 2005). "Early work on the ubiquitin proteasome system, an interview with Aaron Ciechanover. Interview by CDD". Cell Death and Differentiation. 12 (9): 1167–77. doi:10.1038/sj.cdd.4401691. PMID 16094393.
- ↑ Wilk S, Orlowski M (Nov 1980). "Cation-sensitive neutral endopeptidase: isolation and specificity of the bovine pituitary enzyme". Journal of Neurochemistry. 35 (5): 1172–82. doi:10.1111/j.1471-4159.1980.tb07873.x. PMID 6778972.
- ↑ Tanaka K, Waxman L, Goldberg AL (Jun 1983). "ATP serves two distinct roles in protein degradation in reticulocytes, one requiring and one independent of ubiquitin". The Journal of Cell Biology. 96 (6): 1580–5. doi:10.1083/jcb.96.6.1580. PMC 2112434. PMID 6304111.
- ↑ Hough R, Pratt G, Rechsteiner M (Jun 1987). "Purification of two high molecular weight proteases from rabbit reticulocyte lysate". The Journal of Biological Chemistry. 262 (17): 8303–13. PMID 3298229.
- ↑ Hershko A (Sep 2005). "Early work on the ubiquitin proteasome system, an interview with Avram Hershko. Interview by CDD". Cell Death and Differentiation. 12 (9): 1158–61. doi:10.1038/sj.cdd.4401709. PMID 16094391.
- ↑ Kopp F, Steiner R, Dahlmann B, Kuehn L, Reinauer H (Aug 1986). "Size and shape of the multicatalytic proteinase from rat skeletal muscle". Biochimica et Biophysica Acta. 872 (3): 253–60. doi:10.1016/0167-4838(86)90278-5. PMID 3524688.
- ↑ Löwe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R (Apr 1995). "Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution". Science. 268 (5210): 533–9. doi:10.1126/science.7725097. PMID 7725097.
- 1 2 3 4 Wang J, Maldonado MA (Aug 2006). "The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases". Cellular & Molecular Immunology. 3 (4): 255–61. PMID 16978533.
- 1 2 3 Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL (Sep 2007). "Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry". Molecular Cell. 27 (5): 731–44. doi:10.1016/j.molcel.2007.06.033. PMC 2083707. PMID 17803938.
- ↑ Wilk S, Orlowski M (Mar 1983). "Evidence that pituitary cation-sensitive neutral endopeptidase is a multicatalytic protease complex". Journal of Neurochemistry. 40 (3): 842–9. doi:10.1111/j.1471-4159.1983.tb08056.x. PMID 6338156.
- 1 2 Nandi D, Tahiliani P, Kumar A, Chandu D (Mar 2006). "The ubiquitin-proteasome system". Journal of Biosciences. 31 (1): 137–55. doi:10.1007/BF02705243. PMID 16595883.
- 1 2 Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH (Oct 1997). "The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing". The Journal of Biological Chemistry. 272 (40): 25200–9. doi:10.1074/jbc.272.40.25200. PMID 9312134.
- 1 2 Padmanabhan A, Vuong SA, Hochstrasser M (March 2016). "Assembly of an Evolutionarily Conserved Alternative Proteasome Isoform in Human Cells". Cell Reports. 14 (12): 2962–74. doi:10.1016/j.celrep.2016.02.068. PMID 26997268.
- ↑ Velichutina I, Connerly PL, Arendt CS, Li X, Hochstrasser M (February 2004). "Plasticity in eucaryotic 20S proteasome ring assembly revealed by a subunit deletion in yeast". The EMBO Journal. 23 (3): 500–10. doi:10.1038/sj.emboj.7600059. PMID 14739934.
- ↑ Kusmierczyk AR, Kunjappu MJ, Funakoshi M, Hochstrasser M (March 2008). "A multimeric assembly factor controls the formation of alternative 20S proteasomes". Nature Structural & Molecular Biology. 15 (3): 237–44. doi:10.1038/nsmb.1389. PMID 18278055.
- ↑ Zwickl P, Ng D, Woo KM, Klenk HP, Goldberg AL (Sep 1999). "An archaebacterial ATPase, homologous to ATPases in the eukaryotic 26 S proteasome, activates protein breakdown by 20 S proteasomes". The Journal of Biological Chemistry. 274 (37): 26008–14. doi:10.1074/jbc.274.37.26008. PMID 10473546.
- 1 2 3 4 Smith DM, Kafri G, Cheng Y, Ng D, Walz T, Goldberg AL (Dec 2005). "ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins". Molecular Cell. 20 (5): 687–98. doi:10.1016/j.molcel.2005.10.019. PMID 16337593.
- 1 2 3 4 5 Liu CW, Li X, Thompson D, Wooding K, Chang TL, Tang Z, Yu H, Thomas PJ, DeMartino GN (Oct 2006). "ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome". Molecular Cell. 24 (1): 39–50. doi:10.1016/j.molcel.2006.08.025. PMID 17018291.
- ↑ Lam YA, Lawson TG, Velayutham M, Zweier JL, Pickart CM (Apr 2002). "A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal". Nature. 416 (6882): 763–7. doi:10.1038/416763a. PMID 11961560.
- 1 2 3 Beck F, Unverdorben P, Bohn S, Schweitzer A, Pfeifer G, Sakata E, Nickell S, Plitzko JM, Villa E, Baumeister W, Förster F (Sep 2012). "Near-atomic resolution structural model of the yeast 26S proteasome". Proceedings of the National Academy of Sciences of the United States of America. 109 (37): 14870–5. doi:10.1073/pnas.1213333109. PMID 22927375.
- 1 2 Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A (Feb 2012). "Complete subunit architecture of the proteasome regulatory particle". Nature. 482 (7384). doi:10.1038/nature10774. PMC 3285539. PMID 22237024.
- ↑ Lasker K, Förster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W (Jan 2012). "Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach". Proceedings of the National Academy of Sciences of the United States of America. 109 (5): 1380–7. doi:10.1073/pnas.1120559109. PMC 3277140. PMID 22307589.
- 1 2 3 Unverdorben P, Beck F, Śledź P, Schweitzer A, Pfeifer G, Plitzko JM, Baumeister W, Förster F (Apr 2014). "Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome". Proceedings of the National Academy of Sciences of the United States of America. 111 (15): 5544–9. doi:10.1073/pnas.1403409111. PMC 3992697. PMID 24706844.
- ↑ Śledź P, Unverdorben P, Beck F, Pfeifer G, Schweitzer A, Förster F, Baumeister W (Apr 2013). "Structure of the 26S proteasome with ATP-γS bound provides insights into the mechanism of nucleotide-dependent substrate translocation". Proceedings of the National Academy of Sciences of the United States of America. 110 (18): 7264–7269. doi:10.1073/pnas.1305782110. PMID 23589842.
- ↑ Matyskiela ME, Lander GC, Martin A (Jul 2013). "Conformational switching of the 26S proteasome enables substrate degradation". Nature Structural & Molecular Biology. 20 (7): 781–788. doi:10.1038/nsmb.2616. PMC 3712289. PMID 23770819.
- ↑ Köhler A, Cascio P, Leggett DS, Woo KM, Goldberg AL, Finley D (Jun 2001). "The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release". Molecular Cell. 7 (6): 1143–52. doi:10.1016/S1097-2765(01)00274-X. PMID 11430818.
- ↑ Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y (May 2008). "Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases". Molecular Cell. 30 (3): 360–8. doi:10.1016/j.molcel.2008.03.004. PMC 4141531. PMID 18471981.
- ↑ Förster A, Masters EI, Whitby FG, Robinson H, Hill CP (May 2005). "The 1.9 A structure of a proteasome-11S activator complex and implications for proteasome-PAN/PA700 interactions". Molecular Cell. 18 (5): 589–99. doi:10.1016/j.molcel.2005.04.016. PMID 15916965.
- ↑ Witt S, Kwon YD, Sharon M, Felderer K, Beuttler M, Robinson CV, Baumeister W, Jap BK (Jul 2006). "Proteasome assembly triggers a switch required for active-site maturation". Structure. 14 (7): 1179–88. doi:10.1016/j.str.2006.05.019. PMID 16843899.
- ↑ Krüger E, Kloetzel PM, Enenkel C (2001). "20S proteasome biogenesis". Biochimie. 83 (3-4): 289–93. doi:10.1016/S0300-9084(01)01241-X. PMID 11295488.
- ↑ Murata S, Yashiroda H, Tanaka K (Feb 2009). "Molecular mechanisms of proteasome assembly". Nature Reviews Molecular Cell Biology. 10 (2): 104–115. doi:10.1038/nrm2630. PMID 19165213.
- ↑ Sakata E, Stengel F, Fukunaga K, Zhou M, Saeki Y, Förster F, Baumeister W, Tanaka K, Robinson CV (Jun 2011). "The catalytic activity of Ubp6 enhances maturation of the proteasomal regulatory particle". Molecular Cell. 42 (5): 637–649. doi:10.1016/j.molcel.2011.04.021. PMID 21658604.
- ↑ Fukunaga K, Kudo T, Toh-e A, Tanaka K, Saeki Y (Jun 2010). "Dissection of the assembly pathway of the proteasome lid in Saccharomyces cerevisiae". Biochemical and Biophysical Research Communications. 396 (4): 1048–1053. doi:10.1016/j.bbrc.2010.05.061. PMID 20471955.
- ↑ Haas AL, Warms JV, Hershko A, Rose IA (Mar 1982). "Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation". The Journal of Biological Chemistry. 257 (5): 2543–8. PMID 6277905.
- ↑ Thrower JS, Hoffman L, Rechsteiner M, Pickart CM (Jan 2000). "Recognition of the polyubiquitin proteolytic signal". The EMBO Journal. 19 (1): 94–102. doi:10.1093/emboj/19.1.94. PMC 1171781. PMID 10619848.
- ↑ Risseeuw EP, Daskalchuk TE, Banks TW, Liu E, Cotelesage J, Hellmann H, Estelle M, Somers DE, Crosby WL (Jun 2003). "Protein interaction analysis of SCF ubiquitin E3 ligase subunits from Arabidopsis". The Plant Journal. 34 (6): 753–67. doi:10.1046/j.1365-313X.2003.01768.x. PMID 12795696.
- ↑ Elsasser S, Finley D (Aug 2005). "Delivery of ubiquitinated substrates to protein-unfolding machines". Nature Cell Biology. 7 (8): 742–9. doi:10.1038/ncb0805-742. PMID 16056265.
- ↑ Sadanandom A, Bailey M, Ewan R, Lee J, Nelis S (Oct 2012). "The ubiquitin-proteasome system: central modifier of plant signalling". The New Phytologist. 196 (1): 13–28. doi:10.1111/j.1469-8137.2012.04266.x. PMID 22897362.
- ↑ Sharp PM, Li WH (1987). "Ubiquitin genes as a paradigm of concerted evolution of tandem repeats". Journal of Molecular Evolution. 25 (1): 58–64. doi:10.1007/BF02100041. PMID 3041010.
- ↑ Pickart CM, Fushman D (Dec 2004). "Polyubiquitin chains: polymeric protein signals". Current Opinion in Chemical Biology. 8 (6): 610–16. doi:10.1016/j.cbpa.2004.09.009. PMID 15556404.
- ↑ Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J (Apr 2009). "Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation". Cell. 137 (1): 133–45. doi:10.1016/j.cell.2009.01.041. PMC 2668214. PMID 19345192.
- ↑ Pickart CM (Nov 2000). "Ubiquitin in chains". Trends in Biochemical Sciences. 25 (11): 544–8. doi:10.1016/S0968-0004(00)01681-9. PMID 11084366.
- ↑ Zhu Q, Wani G, Wang QE, El-mahdy M, Snapka RM, Wani AA (Jul 2005). "Deubiquitination by proteasome is coordinated with substrate translocation for proteolysis in vivo". Experimental Cell Research. 307 (2): 436–51. doi:10.1016/j.yexcr.2005.03.031. PMID 15950624.
- ↑ Wenzel T, Baumeister W (Mar 1995). "Conformational constraints in protein degradation by the 20S proteasome". Nature Structural Biology. 2 (3): 199–204. doi:10.1038/nsb0395-199. PMID 7773788.
- ↑ Inobe T, Fishbain S, Prakash S, Matouschek A (Mar 2011). "Defining the geometry of the two-component proteasome degron". Nature Chemical Biology. 7 (3): 161–7. doi:10.1038/nchembio.521. PMC 3129032. PMID 21278740.
- ↑ van der Lee R, Lang B, Kruse K, Gsponer J, Sánchez de Groot N, Huynen MA, Matouschek A, Fuxreiter M, Babu MM (Sep 2014). "Intrinsically disordered segments affect protein half-life in the cell and during evolution". Cell Reports. 8 (6): 1832–44. doi:10.1016/j.celrep.2014.07.055. PMC 4358326. PMID 25220455.
- ↑ Smith DM, Benaroudj N, Goldberg A (Oct 2006). "Proteasomes and their associated ATPases: a destructive combination". Journal of Structural Biology. 156 (1): 72–83. doi:10.1016/j.jsb.2006.04.012. PMID 16919475.
- ↑ Hoyt MA, Zich J, Takeuchi J, Zhang M, Govaerts C, Coffino P (Apr 2006). "Glycine-alanine repeats impair proper substrate unfolding by the proteasome". The EMBO Journal. 25 (8): 1720–9. doi:10.1038/sj.emboj.7601058. PMC 1440830. PMID 16601692.
- ↑ Zhang M, Coffino P (Mar 2004). "Repeat sequence of Epstein–Barr virus-encoded nuclear antigen 1 protein interrupts proteasome substrate processing". The Journal of Biological Chemistry. 279 (10): 8635–41. doi:10.1074/jbc.M310449200. PMID 14688254.
- ↑ Voges D, Zwickl P, Baumeister W (1999). "The 26S proteasome: a molecular machine designed for controlled proteolysis". Annual Review of Biochemistry. 68 (1): 1015–68. doi:10.1146/annurev.biochem.68.1.1015. PMID 10872471.
- 1 2 Rape M, Jentsch S (May 2002). "Taking a bite: proteasomal protein processing". Nature Cell Biology. 4 (5): E113–6. doi:10.1038/ncb0502-e113. PMID 11988749.
- ↑ Rape M, Jentsch S (Nov 2004). "Productive RUPture: activation of transcription factors by proteasomal processing". Biochimica et Biophysica Acta. 1695 (1-3): 209–13. doi:10.1016/j.bbamcr.2004.09.022. PMID 15571816.
- ↑ Asher G, Reuven N, Shaul Y (Aug 2006). "20S proteasomes and protein degradation "by default"". BioEssays. 28 (8): 844–9. doi:10.1002/bies.20447. PMID 16927316.
- ↑ Zhang M, Pickart CM, Coffino P (Apr 2003). "Determinants of proteasome recognition of ornithine decarboxylase, a ubiquitin-independent substrate". The EMBO Journal. 22 (7): 1488–96. doi:10.1093/emboj/cdg158. PMC 152902. PMID 12660156.
- ↑ Asher G, Shaul Y (Aug 2005). "p53 proteasomal degradation: poly-ubiquitination is not the whole story". Cell Cycle. 4 (8): 1015–8. doi:10.4161/cc.4.8.1900. PMID 16082197.
- 1 2 Shringarpure R, Grune T, Mehlhase J, Davies KJ (Jan 2003). "Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome". The Journal of Biological Chemistry. 278 (1): 311–8. doi:10.1074/jbc.M206279200. PMID 12401807.
- 1 2 3 Gille C, Goede A, Schlöetelburg C, Preissner R, Kloetzel PM, Göbel UB, Frömmel C (Mar 2003). "A comprehensive view on proteasomal sequences: implications for the evolution of the proteasome". Journal of Molecular Biology. 326 (5): 1437–48. doi:10.1016/S0022-2836(02)01470-5. PMID 12595256.
- ↑ Bochtler M, Ditzel L, Groll M, Hartmann C, Huber R (1999). "The proteasome". Annual Review of Biophysics and Biomolecular Structure. 28 (1): 295–317. doi:10.1146/annurev.biophys.28.1.295. PMID 10410804.
- ↑ Chesnel F, Bazile F, Pascal A, Kubiak JZ (Aug 2006). "Cyclin B dissociation from CDK1 precedes its degradation upon MPF inactivation in mitotic extracts of Xenopus laevis embryos". Cell Cycle. 5 (15): 1687–98. doi:10.4161/cc.5.15.3123. PMID 16921258.
- ↑ Brito DA, Rieder CL (Jun 2006). "Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint". Current Biology. 16 (12): 1194–200. doi:10.1016/j.cub.2006.04.043. PMC 2749311. PMID 16782009.
- ↑ Havens CG, Ho A, Yoshioka N, Dowdy SF (Jun 2006). "Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species". Molecular and Cellular Biology. 26 (12): 4701–11. doi:10.1128/MCB.00303-06. PMC 1489138. PMID 16738333.
- ↑ Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M (Mar 2004). "Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase". Nature. 428 (6979): 190–3. doi:10.1038/nature02330. PMID 15014502.
- ↑ Higashitsuji H, Liu Y, Mayer RJ, Fujita J (Oct 2005). "The oncoprotein gankyrin negatively regulates both p53 and RB by enhancing proteasomal degradation". Cell Cycle. 4 (10): 1335–7. doi:10.4161/cc.4.10.2107. PMID 16177571.
- ↑ Dharmasiri S, Estelle M (2002). "The role of regulated protein degradation in auxin response". Plant Molecular Biology. 49 (3-4): 401–9. doi:10.1023/A:1015203013208. PMID 12036263.
- ↑ Weijers D, Benkova E, Jäger KE, Schlereth A, Hamann T, Kientz M, Wilmoth JC, Reed JW, Jürgens G (May 2005). "Developmental specificity of auxin response by pairs of ARF and Aux/IAA transcriptional regulators". The EMBO Journal. 24 (10): 1874–85. doi:10.1038/sj.emboj.7600659. PMC 1142592. PMID 15889151.
- ↑ Haas AL, Baboshina O, Williams B, Schwartz LM (Apr 1995). "Coordinated induction of the ubiquitin conjugation pathway accompanies the developmentally programmed death of insect skeletal muscle". The Journal of Biological Chemistry. 270 (16): 9407–12. doi:10.1074/jbc.270.16.9407. PMID 7721865.
- ↑ Schwartz LM, Myer A, Kosz L, Engelstein M, Maier C (Oct 1990). "Activation of polyubiquitin gene expression during developmentally programmed cell death". Neuron. 5 (4): 411–9. doi:10.1016/0896-6273(90)90080-Y. PMID 2169771.
- ↑ Löw P, Bussell K, Dawson SP, Billett MA, Mayer RJ, Reynolds SE (Jan 1997). "Expression of a 26S proteasome ATPase subunit, MS73, in muscles that undergo developmentally programmed cell death, and its control by ecdysteroid hormones in the insect Manduca sexta". FEBS Letters. 400 (3): 345–9. doi:10.1016/S0014-5793(96)01413-5. PMID 9009228.
- ↑ Pitzer F, Dantes A, Fuchs T, Baumeister W, Amsterdam A (Sep 1996). "Removal of proteasomes from the nucleus and their accumulation in apoptotic blebs during programmed cell death". FEBS Letters. 394 (1): 47–50. doi:10.1016/0014-5793(96)00920-9. PMID 8925925.
- 1 2 Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ (Jun 1999). "Proteasome inhibitors: a novel class of potent and effective antitumor agents". Cancer Research. 59 (11): 2615–22. PMID 10363983.
- ↑ Orlowski RZ (Apr 1999). "The role of the ubiquitin-proteasome pathway in apoptosis". Cell Death and Differentiation. 6 (4): 303–13. doi:10.1038/sj.cdd.4400505. PMID 10381632.
- ↑ Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G (Nov 2006). "Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties". Cell Cycle. 5 (22): 2592–601. doi:10.4161/cc.5.22.3448. PMID 17106261.
- ↑ Park SH, Bolender N, Eisele F, Kostova Z, Takeuchi J, Coffino P, Wolf DH (Jan 2007). "The cytoplasmic Hsp70 chaperone machinery subjects misfolded and endoplasmic reticulum import-incompetent proteins to degradation via the ubiquitin-proteasome system". Molecular Biology of the Cell. 18 (1): 153–65. doi:10.1091/mbc.E06-04-0338. PMC 1751312. PMID 17065559.
- ↑ Dai Q, Qian SB, Li HH, McDonough H, Borchers C, Huang D, Takayama S, Younger JM, Ren HY, Cyr DM, Patterson C (Nov 2005). "Regulation of the cytoplasmic quality control protein degradation pathway by BAG2". The Journal of Biological Chemistry. 280 (46): 38673–81. doi:10.1074/jbc.M507986200. PMID 16169850.
- ↑ Bader N, Grune T (2006). "Protein oxidation and proteolysis". Biological Chemistry. 387 (10-11): 1351–5. doi:10.1515/BC.2006.169. PMID 17081106.
- ↑ Davies KJ (2003). "Degradation of oxidized proteins by the 20S proteasome". Biochimie. 83 (3-4): 301–10. doi:10.1016/S0300-9084(01)01250-0. PMID 11295490.
- 1 2 Lehman NL (Sep 2009). "The ubiquitin proteasome system in neuropathology". Acta Neuropathologica. 118 (3): 329–47. doi:10.1007/s00401-009-0560-x. PMC 2716447. PMID 19597829.
- ↑ McNaught KS, Jackson T, JnoBaptiste R, Kapustin A, Olanow CW (May 2006). "Proteasomal dysfunction in sporadic Parkinson's disease". Neurology. 66 (10 Suppl 4): S37–49. doi:10.1212/01.wnl.0000221745.58886.2e. PMID 16717251.
- ↑ Sharma N, Brandis KA, Herrera SK, Johnson BE, Vaidya T, Shrestha R, Debburman SK (2006). "alpha-Synuclein budding yeast model: toxicity enhanced by impaired proteasome and oxidative stress". Journal of Molecular Neuroscience. 28 (2): 161–78. doi:10.1385/JMN:28:2:161. PMID 16679556.
- ↑ Murata S, Sasaki K, Kishimoto T, Niwa S, Hayashi H, Takahama Y, Tanaka K (Jun 2007). "Regulation of CD8+ T cell development by thymus-specific proteasomes". Science. 316 (5829): 1349–53. doi:10.1126/science.1141915. PMID 17540904.
- ↑ Cascio P, Hilton C, Kisselev AF, Rock KL, Goldberg AL (May 2001). "26S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide". The EMBO Journal. 20 (10): 2357–66. doi:10.1093/emboj/20.10.2357. PMC 125470. PMID 11350924.
- ↑ Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC (Nov 2010). "Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21)". Proceedings of the National Academy of Sciences of the United States of America. 107 (46): 19985–19990. doi:10.1073/pnas.1014074107. PMC 2993423. PMID 21045130.
- ↑ Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL (May 1995). "Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin". Science. 268 (5211): 726–31. doi:10.1126/science.7732382. PMID 7732382.
- ↑ United States Food and Drug Administration press release 13 May 2003. Access date 29 December 2006. See also FDA Velcade information page.
- ↑ Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Stadtmauer EA, O'Connor OA, Shi H, Boral AL, Goy A (Oct 2006). "Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma". Journal of Clinical Oncology. 24 (30): 4867–74. doi:10.1200/JCO.2006.07.9665. PMID 17001068.
- ↑ Jakob C, Egerer K, Liebisch P, Türkmen S, Zavrski I, Kuckelkorn U, Heider U, Kaiser M, Fleissner C, Sterz J, Kleeberg L, Feist E, Burmester GR, Kloetzel PM, Sezer O (Mar 2007). "Circulating proteasome levels are an independent prognostic factor for survival in multiple myeloma". Blood. 109 (5): 2100–5. doi:10.1182/blood-2006-04-016360. PMID 17095627.
- ↑ Shah SA, Potter MW, McDade TP, Ricciardi R, Perugini RA, Elliott PJ, Adams J, Callery MP (2001). "26S proteasome inhibition induces apoptosis and limits growth of human pancreatic cancer". Journal of Cellular Biochemistry. 82 (1): 110–22. doi:10.1002/jcb.1150. PMID 11400168.
- ↑ Nawrocki ST, Sweeney-Gotsch B, Takamori R, McConkey DJ (Jan 2004). "The proteasome inhibitor bortezomib enhances the activity of docetaxel in orthotopic human pancreatic tumor xenografts". Molecular Cancer Therapeutics. 3 (1): 59–70. PMID 14749476.
- ↑ Schenkein D (Jun 2002). "Proteasome inhibitors in the treatment of B-cell malignancies". Clinical Lymphoma. 3 (1): 49–55. doi:10.3816/CLM.2002.n.011. PMID 12141956.
- ↑ O'Connor OA, Wright J, Moskowitz C, Muzzy J, MacGregor-Cortelli B, Stubblefield M, Straus D, Portlock C, Hamlin P, Choi E, Dumetrescu O, Esseltine D, Trehu E, Adams J, Schenkein D, Zelenetz AD (Feb 2005). "Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin's lymphoma and mantle cell lymphoma". Journal of Clinical Oncology. 23 (4): 676–84. doi:10.1200/JCO.2005.02.050. PMID 15613699.
- ↑ Messinger YH, Gaynon PS, Sposto R, van der Giessen J, Eckroth E, Malvar J, Bostrom BC (Jul 2012). "Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study". Blood. 120 (2): 285–90. doi:10.1182/blood-2012-04-418640. PMID 22653976.
- ↑ Lambrou GI, Papadimitriou L, Chrousos GP, Vlahopoulos SA (Apr 2012). "Glucocorticoid and proteasome inhibitor impact on the leukemic lymphoblast: multiple, diverse signals converging on a few key downstream regulators". Molecular and Cellular Endocrinology. 351 (2): 142–51. doi:10.1016/j.mce.2012.01.003. PMID 22273806.
- ↑ Schmidtke G, Holzhütter HG, Bogyo M, Kairies N, Groll M, de Giuli R, Emch S, Groettrup M (Dec 1999). "How an inhibitor of the HIV-I protease modulates proteasome activity". The Journal of Biological Chemistry. 274 (50): 35734–40. doi:10.1074/jbc.274.50.35734. PMID 10585454.
- ↑ Laurent N, de Boüard S, Guillamo JS, Christov C, Zini R, Jouault H, Andre P, Lotteau V, Peschanski M (Feb 2004). "Effects of the proteasome inhibitor ritonavir on glioma growth in vitro and in vivo". Molecular Cancer Therapeutics. 3 (2): 129–36. PMID 14985453.
- ↑ Zollner TM, Podda M, Pien C, Elliott PJ, Kaufmann R, Boehncke WH (Mar 2002). "Proteasome inhibition reduces superantigen-mediated T cell activation and the severity of psoriasis in a SCID-hu model". The Journal of Clinical Investigation. 109 (5): 671–9. doi:10.1172/JCI12736. PMC 150886. PMID 11877475.
- ↑ Elliott PJ, Pien CS, McCormack TA, Chapman ID, Adams J (Aug 1999). "Proteasome inhibition: A novel mechanism to combat asthma". The Journal of Allergy and Clinical Immunology. 104 (2 Pt 1): 294–300. doi:10.1016/S0091-6749(99)70369-6. PMID 10452747.
- ↑ Verdoes M, Florea BI, Menendez-Benito V, Maynard CJ, Witte MD, van der Linden WA, van den Nieuwendijk AM, Hofmann T, Berkers CR, van Leeuwen FW, Groothuis TA, Leeuwenburgh MA, Ovaa H, Neefjes JJ, Filippov DV, van der Marel GA, Dantuma NP, Overkleeft HS (Nov 2006). "A fluorescent broad-spectrum proteasome inhibitor for labeling proteasomes in vitro and in vivo". Chemistry & Biology. 13 (11): 1217–26. doi:10.1016/j.chembiol.2006.09.013. PMID 17114003.
- ↑ Kleiger G, Mayor T (Jun 2014). "Perilous journey: a tour of the ubiquitin-proteasome system". Trends in Cell Biology. 24 (6): 352–9. doi:10.1016/j.tcb.2013.12.003. PMC 4037451. PMID 24457024.
- ↑ Goldberg AL, Stein R, Adams J (Aug 1995). "New insights into proteasome function: from archaebacteria to drug development". Chemistry & Biology. 2 (8): 503–8. doi:10.1016/1074-5521(95)90182-5. PMID 9383453.
- ↑ Sulistio YA, Heese K (Jan 2015). "The Ubiquitin-Proteasome System and Molecular Chaperone Deregulation in Alzheimer's Disease". Molecular Neurobiology. doi:10.1007/s12035-014-9063-4. PMID 25561438.
- ↑ Ortega Z, Lucas JJ (2014). "Ubiquitin-proteasome system involvement in Huntington's disease". Frontiers in Molecular Neuroscience. 7: 77. doi:10.3389/fnmol.2014.00077. PMC 4179678. PMID 25324717.
- ↑ Sandri M, Robbins J (Jun 2014). "Proteotoxicity: an underappreciated pathology in cardiac disease". Journal of Molecular and Cellular Cardiology. 71: 3–10. doi:10.1016/j.yjmcc.2013.12.015. PMC 4011959. PMID 24380730.
- ↑ Drews O, Taegtmeyer H (Dec 2014). "Targeting the ubiquitin-proteasome system in heart disease: the basis for new therapeutic strategies". Antioxidants & Redox Signaling. 21 (17): 2322–43. doi:10.1089/ars.2013.5823. PMC 4241867. PMID 25133688.
- ↑ Wang ZV, Hill JA (Feb 2015). "Protein quality control and metabolism: bidirectional control in the heart". Cell Metabolism. 21 (2): 215–26. doi:10.1016/j.cmet.2015.01.016. PMC 4317573. PMID 25651176.
- 1 2 Karin M, Delhase M (Feb 2000). "The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling". Seminars in Immunology. 12 (1): 85–98. doi:10.1006/smim.2000.0210. PMID 10723801.
- ↑ Ermolaeva MA, Dakhovnik A, Schumacher B (Jan 2015). "Quality control mechanisms in cellular and systemic DNA damage responses". Ageing Research Reviews. 23: 3–11. doi:10.1016/j.arr.2014.12.009. PMID 25560147.
- ↑ Checler F, da Costa CA, Ancolio K, Chevallier N, Lopez-Perez E, Marambaud P (Jul 2000). "Role of the proteasome in Alzheimer's disease". Biochimica et Biophysica Acta. 1502 (1): 133–8. doi:10.1016/s0925-4439(00)00039-9. PMID 10899438.
- 1 2 Chung KK, Dawson VL, Dawson TM (Nov 2001). "The role of the ubiquitin-proteasomal pathway in Parkinson's disease and other neurodegenerative disorders". Trends in Neurosciences. 24 (11 Suppl): S7–14. doi:10.1016/s0166-2236(00)01998-6. PMID 11881748.
- 1 2 Ikeda K, Akiyama H, Arai T, Ueno H, Tsuchiya K, Kosaka K (Jul 2002). "Morphometrical reappraisal of motor neuron system of Pick's disease and amyotrophic lateral sclerosis with dementia". Acta Neuropathologica. 104 (1): 21–8. doi:10.1007/s00401-001-0513-5. PMID 12070660.
- ↑ Manaka H, Kato T, Kurita K, Katagiri T, Shikama Y, Kujirai K, Kawanami T, Suzuki Y, Nihei K, Sasaki H (May 1992). "Marked increase in cerebrospinal fluid ubiquitin in Creutzfeldt–Jakob disease". Neuroscience Letters. 139 (1): 47–9. doi:10.1016/0304-3940(92)90854-z. PMID 1328965.
- ↑ Mathews KD, Moore SA (Jan 2003). "Limb-girdle muscular dystrophy". Current Neurology and Neuroscience Reports. 3 (1): 78–85. doi:10.1007/s11910-003-0042-9. PMID 12507416.
- ↑ Mayer RJ (Mar 2003). "From neurodegeneration to neurohomeostasis: the role of ubiquitin". Drug News & Perspectives. 16 (2): 103–8. doi:10.1358/dnp.2003.16.2.829327. PMID 12792671.
- ↑ Calise J, Powell SR (Feb 2013). "The ubiquitin proteasome system and myocardial ischemia". American Journal of Physiology. Heart and Circulatory Physiology. 304 (3): H337–49. doi:10.1152/ajpheart.00604.2012. PMC 3774499. PMID 23220331.
- ↑ Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM (Mar 2010). "Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies". Circulation. 121 (8): 997–1004. doi:10.1161/CIRCULATIONAHA.109.904557. PMC 2857348. PMID 20159828.
- ↑ Powell SR (Jul 2006). "The ubiquitin-proteasome system in cardiac physiology and pathology". American Journal of Physiology. Heart and Circulatory Physiology. 291 (1): H1–H19. doi:10.1152/ajpheart.00062.2006. PMID 16501026.
- ↑ Adams J (Apr 2003). "Potential for proteasome inhibition in the treatment of cancer". Drug Discovery Today. 8 (7): 307–15. doi:10.1016/s1359-6446(03)02647-3. PMID 12654543.
- ↑ Ben-Neriah Y (Jan 2002). "Regulatory functions of ubiquitination in the immune system". Nature Immunology. 3 (1): 20–6. doi:10.1038/ni0102-20. PMID 11753406.
- ↑ Egerer K, Kuckelkorn U, Rudolph PE, Rückert JC, Dörner T, Burmester GR, Kloetzel PM, Feist E (Oct 2002). "Circulating proteasomes are markers of cell damage and immunologic activity in autoimmune diseases". The Journal of Rheumatology. 29 (10): 2045–52. PMID 12375310.
Further reading
- Glickman MH, Adir N (Jan 2004). "The proteasome and the delicate balance between destruction and rescue". PLoS Biology. 2 (1): e13. doi:10.1371/journal.pbio.0020013. PMC 314468. PMID 14737189.
- The Yeast 26S Proteasome with list of subunits and pictures
- Ciechanover A (Sep 2005). "Early work on the ubiquitin proteasome system, an interview with Aaron Ciechanover. Interview by CDD". Cell Death and Differentiation. 12 (9): 1167–77. doi:10.1038/sj.cdd.4401691. PMID 16094393.
- Hershko A (Sep 2005). "Early work on the ubiquitin proteasome system, an interview with Avram Hershko. Interview by CDD". Cell Death and Differentiation. 12 (9): 1158–61. doi:10.1038/sj.cdd.4401709. PMID 16094391.
- Rose I (Sep 2005). "Early work on the ubiquitin proteasome system, an interview with Irwin Rose. Interview by CDD". Cell Death and Differentiation. 12 (9): 1162–6. doi:10.1038/sj.cdd.4401700. PMID 16094392.
- Cvek B, Dvorak Z (2007). "Targeting of nuclear factor-kappaB and proteasome by dithiocarbamate complexes with metals". Current Pharmaceutical Design. 13 (30): 3155–67. doi:10.2174/138161207782110390. PMID 17979756.
External links
| Wikimedia Commons has media related to Proteasomes. |