Fragile X syndrome
| Fragile X syndrome | |
|---|---|
| Synonyms |
Martin-Bell syndrome,[1] Escalante syndrome |
 | |
| Boy with fragile X syndrome | |
| Classification and external resources | |
| Specialty | Medical genetics, pediatrics, psychiatry |
| ICD-10 | Q99.2 |
| ICD-9-CM | 759.83 |
| OMIM | 300624 |
| DiseasesDB | 4973 |
| MedlinePlus | 001668 |
| eMedicine | ped/800 |
| Patient UK | Fragile X syndrome |
| MeSH | D005600 |
| Orphanet | 908 |
Fragile X syndrome (FXS) is a genetic disorder. Symptoms often include mild to moderate intellectual disability. Physical features may include a long and narrow face, large ears, flexible fingers, and large testicles. About a third of people have features of autism such as problems with social interactions and delayed speech. Hyperactivity is common and seizures occur in about 10%. Males are usually more affected than females.[1]
Fragile X syndrome is typically due to the expansion of the CGG triplet repeat within the Fragile X mental retardation 1 (FMR1) gene on the X chromosome.[1] This results in a failure to express the fragile X mental retardation protein (FMRP), which is required for normal neural development. Depending on the length of the CGG repeat, an allele may be classified as normal (unaffected by the syndrome), a premutation (at risk of fragile X associated disorders), or full mutation (usually affected by the syndrome).[2] A diagnosis of fragile X syndrome is made through genetic testing to determine the number of CGG repeats. Testing for premutation carriers may allow for genetic counseling.
There is no medication that has a benefit specifically for fragile X syndrome. Medications are commonly used to treat symptoms of attention deficit and hyperactivity, anxiety, and aggression. Supportive management is important in optimizing functioning in individuals with fragile X syndrome, and may involve speech therapy, occupational therapy, and individualized educational and behavioral programs. Early intervention is recommended as it provides the most opportunity for developing a full range of skills.[3] These interventions include education and medications with accompanied therapies.[3]
Fragile X syndrome occurs in about 1 in 4,000 males and 1 in 8,000 females.[1] The first complete DNA sequence of the repeat expansion in someone with the full mutation was generated by scientists in 2012 using SMRT sequencing.[4]
Signs and symptoms

Most young children do not show any physical signs of FXS. [5] It is not until puberty that physical features of FXS begin to develop. [5] Aside from intellectual disability, prominent characteristics of the syndrome may include an elongated face, large or protruding ears, flat feet, larger testes (macroorchidism), and low muscle tone.[6][7] Recurrent otitis media (middle ear infection) and sinusitis is common during early childhood. Speech may be cluttered or nervous. Behavioral characteristics may include stereotypic movements (e.g., hand-flapping) and atypical social development, particularly shyness, limited eye contact, memory problems, and difficulty with face encoding. Some individuals with fragile X syndrome also meet the diagnostic criteria for autism.
Males with a full mutation display virtually complete penetrance and will therefore almost always display symptoms of FXS, while females with a full mutation generally display a penetrance of about 50% as a result of having a second, normal X chromosome.[8] Females with FXS may have symptoms ranging from mild to severe, although they are generally less affected than males.
Physical phenotype
- Large, protruding ears (both)
- Long face (vertical maxillary excess)
- High-arched palate (related to the above)
- Hyperextensible finger joints
- Hyperextensible ('Double-jointed') thumbs
- Flat feet
- Soft skin
- Postpubescent macroorchidism (Large testicles in men after puberty)[9]
- Hypotonia (low muscle tone)[10]
Intellectual development
Individuals with FXS may present anywhere on a continuum from learning disabilities in the context of a normal intelligence quotient (IQ) to severe intellectual disability, with an average IQ of 40 in males who have complete silencing of the FMR1 gene.[7] Females, who tend to be less affected, generally have an IQ which is normal or borderline with learning difficulties. The main difficulties in individuals with FXS are with working and short-term memory, executive function, visual memory, visual-spatial relationships, and mathematics, with verbal abilities being relatively spared.[7][11]
Data on intellectual development in FXS are limited. However, there is some evidence that standardized IQ decreases over time in the majority of cases, apparently as a result of slowed intellectual development. A longitudinal study looking at pairs of siblings where one child was affected and the other was not found that affected children had an intellectual learning rate which was 55% slower than unaffected children.[11]
When both autism and FXS are present, a greater language deficit and lower IQ is observed as compared to children with only FXS.[12]
Autism
Fragile X syndrome co-occurs with autism in many cases and is a suspected genetic cause of the autism in these cases.[6][13] This finding has resulted in screening for FMR1 mutation to be considered mandatory in children diagnosed with autism.[6] Of those with fragile X syndrome, prevalence of concurrent autism spectrum disorder (ASD) has been estimated to be between 15 and 60%, with the variation due to differences in diagnostic methods and the high frequency of autistic features in individuals with fragile X syndrome not meeting the DSM criteria for an ASD.[13]
Although individuals with FXS have difficulties in forming friendships, those with FXS and ASD characteristically also have difficulties with reciprocal conversation with their peers. Social withdrawal behaviors, including avoidance and indifference, appear to be the best predictors of ASD in FXS, with avoidance appearing to be correlated more with social anxiety while indifference was more strongly correlated to severe ASD.[13] When both autism and FXS are present, a greater language deficit and lower IQ is observed as compared to children with only FXS.[12]
Genetic mouse models of FXS have also been shown to have autistic-like behaviors.[14][15][16][17][18]
Social interaction
FXS is characterized by social anxiety, including poor eye contact, gaze aversion, prolonged time to commence social interaction, and challenges forming peer relationships.[19] Social anxiety is one of the most common features associated with FXS, with up to 75% of males in one series characterized as having excessive shyness and 50% having panic attacks.[13] Social anxiety in individuals with FXS is related to challenges with face encoding, the ability to recognize a face that one has seen before.[20]
It appears that individuals with FXS are interested in social interaction and display greater empathy than groups with other causes of intellectual disability, but display anxiety and withdrawal when placed in unfamiliar situations with unfamiliar people.[13][19] This may range from mild social withdrawal, which is predominantly associated with shyness, to severe social withdrawal, which may be associated with co-existing autism spectrum disorder.[13]
Females with FXS frequently display shyness, social anxiety and social avoidance or withdrawal.[7] In addition, premutation in females has been found to be associated with social anxiety.
Individuals with FXS show decreased activation in the prefrontal regions of the brain.
Psychiatric
Attention deficit hyperactivity disorder (ADHD) is found in the majority of males with FXS and 30% of females, making it the most common psychiatric diagnosis in those with FXS.[6][19] Hyperactivity and disruptive behavior peak in the preschool years and then gradually decline with age, although inattentive symptoms are generally lifelong.[19]
Aside from the characteristic social phobia features, a range of other anxiety symptoms are very commonly associated with FXS, with symptoms typically spanning a number of psychiatric diagnoses but not fulfilling any of the criteria in full.[19] Behaviors such as hand flapping and biting, as well as aggression, can be an expression of anxiety. Although only a minority will meet the criteria for obsessive–compulsive disorder (OCD), a significant majority will feature obsessive-type symptoms. However, as individuals with FXS generally find these behaviors pleasurable, unlike individuals with OCD, they are more frequently referred to as stereotypic behaviors.
Mood symptoms in individuals with FXS rarely meet diagnostic criteria for a major mood disorder as they are typically not of sustained duration.[19] Instead, these are usually transient and related to stressors, and may involve labile (fluctuating) mood, irritability, self-injury and aggression.
Individuals with fragile X-associated tremor/ataxia syndrome (FXTAS) are likely to experience combinations of dementia, mood, and anxiety disorders. Males with the FMR1 premutation and clinical evidence of FXTAS were found to have increased occurrence of somatization, obsessive–compulsive disorder, interpersonal sensitivity, depression, phobic anxiety, and psychoticism.[21]
Hypersensitivity and repetitive behavior
Children with fragile X have very short attention spans, are hyperactive, and show hypersensitivity to visual, auditory, tactile, and olfactory stimuli. These children have difficulty in large crowds due to the loud noises and this can lead to tantrums due to hyperarousal. Children with FXS pull away from light touch and can find textures of materials to be irritating. Transitions from one location to another can be difficult for children with FXS. Behavioral therapy can be used to decrease the child’s sensitivity in some cases.[10]
Perseveration is a common communicative and behavioral characteristic in FXS. Children with FXS may repeat a certain ordinary activity over and over. In speech, the trend is not only in repeating the same phrase but also talking about the same subject continually. Cluttered speech and self-talk are commonly seen. Self-talk includes talking with oneself using different tones and pitches.[10]
Vision
Ophthalmologic problems include strabismus. This requires early identification to avoid amblyopia. Surgery or patching are usually necessary to treat strabismus if diagnosed early. Refractive errors in patients with FXS are also common.[12]
Neurological
Individuals with FXS are at a higher risk of developing seizures, with rates between 10% and 40% reported in the literature.[22] In larger study populations the frequency varies between 13% and 18%,[7][22] consistent with a recent survey of caregivers which found that 14% of males and 6% of females experienced seizures.[22] The seizures tend to be partial, are generally not frequent, and are amenable to treatment with medication.
Individuals who are carriers of premutation alleles are at risk for developing fragile X-associated tremor/ataxia syndrome (FXTAS), a progressive neurodegenerative disease.[8][23] It is seen in approximately half of male carriers over the age of 70, while penetrance in females is lower. Typically, onset of tremor occurs in the sixth decade of life, with subsequent progression to ataxia (loss of coordination) and gradual cognitive decline.[23]
Working memory
From their 40s onward, males with FXS begin developing progressively more severe problems in performing tasks that require the central executive of working memory. Working memory involves the temporary storage of information 'in mind', while processing the same or other information. Phonological memory (or verbal working memory) deteriorates with age in males, while visual-spatial memory is not found to be directly related to age. Males often experience an impairment in the functioning of the phonological loop. The CGG length is significantly correlated with central executive and the visual–spatial memory. However, in a premutation individual, CGG length is only significantly correlated with the central executive, not with either phonological memory or visual–spatial memory.[24]
Fertility
About 20% of women who are carriers for the fragile X premutation are affected by fragile X-related primary ovarian insufficiency (FXPOI), which is defined as menopause before the age of 40.[8][23] The number of CGG repeats correlates with penetrance and age of onset.[8] However premature menopause is more common in premutation carriers than in women with the full mutation, and for premutations with more than 100 repeats the risk of FXPOI begins to decrease.[25] Fragile X-associated primary ovarian insufficiency (FXPOI) is one of three Fragile X-associated Disorders (FXD) caused by changes in the FMR1 gene.FXPOI affects female premutation carriers of Fragile X syndrome, which is caused by the FMR1 gene, when their ovaries are not functioning properly. Women with FXPOI may develop menopause-like symptoms but they are not actually menopausal. Women with FXPOI can still get pregnant in some cases because their ovaries occasionally release viable eggs.[26]
Causes
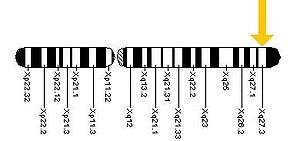
Fragile X syndrome is a genetic disorder which occurs as a result of a mutation of the fragile X mental retardation 1 (FMR1) gene on the X chromosome, most commonly an increase in the number of CGG trinucleotide repeats in the 5' untranslated region of FMR1.[8][23] Mutation at that site is found in 1 out of about every 2000 males and 1 out of about every 259 females. Incidence of the disorder itself is about 1 in every 3600 males and 1 in 4000–6000 females.[27] Although this accounts for over 98% of cases, FXS can also occur as a result of point mutations affecting FMR1.[8][23]
In unaffected individuals, the FMR1 gene contains 5–44 repeats of the sequence CGG, most commonly 29 or 30 repeats.[8][23][28] Between 45 and 54 repeats is considered a "grey zone", with a premutation allele generally considered to be between 55 and 200 repeats in length. Individuals with fragile X syndrome have a full mutation of the FMR1 allele, with over 200 CGG repeats.[6][28][29] In these individuals with a repeat expansion greater than 200, there is methylation of the CGG repeat expansion and FMR1 promoter, leading to the silencing of the FMR1 gene and a lack of its product.
This methylation of FMR1 in chromosome band Xq27.3 is believed to result in constriction of the X chromosome which appears 'fragile' under the microscope at that point, a phenomenon that gave the syndrome its name. One study found that FMR1 silencing is mediated by the FMR1 mRNA. The FMR1 mRNA contains the transcribed CGG-repeat tract as part of the 5' untranslated region, which hybridizes to the complementary CGG-repeat portion of the FMR1 gene to form an RNA·DNA duplex.[30]
Inheritance
Fragile X syndrome has traditionally been considered an X-linked dominant condition with variable expressivity and possibly reduced penetrance.[7] However, due to genetic anticipation and X-inactivation in females, the inheritance of Fragile X syndrome does not follow the usual pattern of X-linked dominant inheritance and some scholars have suggested discontinuing labeling X-linked disorders as dominant or recessive.[31] Females with full FMR1 mutations may have a milder phenotype than males due to variability in X-inactivation.
Before the FMR1 gene was discovered, analysis of pedigrees showed the presence of male carriers who were asymptomatic, with their grandchildren affected by the condition at a higher rate than their siblings suggesting that genetic anticipation was occurring.[8] This tendency for future generations to be affected at a higher frequency became known as the Sherman paradox after its description in 1985.[8][32] Due to this, male children often have a greater degree of symptoms than their mothers.[33]
The explanation for this phenomenon is that male carriers pass on their premutation to all of their daughters, with the length of the FMR1 CGG repeat typically not increasing during meiosis, the cell division that is required to produce sperm.[8][23] Incidentally, males with a full mutation only pass on premutations to their daughters.[23] However, females with a full mutation are able to pass this full mutation on, so theoretically there is a 50% chance that a child will be affected.[23][28] In addition, the length of the CGG repeat frequently does increase during meiosis in female premutation carriers due to instability and so, depending on the length of their premutation, they may pass on a full mutation to their children who will then be affected.
Pathophysiology
FMRP is found throughout the body, but in highest concentrations within the brain and testes.[6][8] It appears to be primarily responsible for selectively binding to around 4% of mRNA in mammalian brains and transporting it out of the cell nucleus and to the synapses of neurons. Most of these mRNA targets have been found to be located in the dendrites of neurons, and brain tissue from humans with FXS and mouse models shows abnormal dendritic spines, which are required to increase contact with other neurons. The subsequent abnormalities in the formation and function of synapses and development of neural circuits result in impaired neuroplasticity, an integral part of memory and learning.[6][8][34] Wiring changes have long been suspected to be involved in the sensory pathophysiology[35] and most recently a range of circuit alterations have been shown, involving structurally increased local connectivity and functionally decreased long-range connectivity.[36]
In addition, FMRP has been implicated in several signalling pathways that are being targeted by a number of drugs undergoing clinical trials. The group 1 metabotropic glutamate receptor (mGluR) pathway, which includes mGluR1 and mGluR5, is involved in mGluR-dependent long term depression (LTD) and long term potentiation (LTP), both of which are important mechanisms in learning.[6][8] The lack of FMRP, which represses mRNA production and thereby protein synthesis, leads to exaggerated LTD. FMRP also appears to affect dopamine pathways in the prefrontal cortex which is believed to result in the attention deficit, hyperactivity and impulse control problems associated with FXS.[6][8][19] The downregulation of GABA pathways, which serve an inhibitory function and are involved in learning and memory, may be a factor in the anxiety symptoms which are commonly seen in FXS.
Diagnosis
Cytogenetic analysis for fragile X syndrome was first available in the late 1970s when diagnosis of the syndrome and carrier status could be determined by culturing cells in a folate deficient medium and then assessing for "fragile sites" (discontinuity of staining in the region of the trinucleotide repeat) on the long arm of the X chromosome.[37] This technique proved unreliable, however, as the fragile site was often seen in less than 40% of an individual's cells. This was not as much of a problem in males, but in female carriers, where the fragile site could generally only be seen in 10% of cells, the mutation often could not be visualised.
Since the 1990s, more sensitive molecular techniques have been used to determine carrier status.[37] The fragile X abnormality is now directly determined by analysis of the number of CGG repeats using polymerase chain reaction (PCR) and methylation status using Southern blot analysis.[7] By determining the number of CGG repeats on the X chromosome, this method allows for more accurate assessment of risk for premutation carriers in terms of their own risk of fragile X associated syndromes, as well as their risk of having affected children. Because this method only tests for expansion of the CGG repeat, individuals with FXS due to missense mutations or deletions involving FMR1 will not be diagnosed using this test and should therefore undergo sequencing of the FMR1 gene if there is clinical suspicion of FXS.
Prenatal testing with chorionic villus sampling or amniocentesis allows diagnosis of FMR1 mutation while the fetus is in utero and appears to be reliable.[7]
Early diagnosis of fragile X syndrome or carrier status is important for providing early intervention in children or fetuses with the syndrome, and allowing genetic counselling with regards to the potential for a couple's future children to be affected. Most parents notice delays in speech and language skills, difficulties in social and emotional domains as well as sensitivity levels in certain situations with their children. [38]
Management
There are no current treatments or cures for the underlying defects of FXS. Management of FXS may include speech therapy, behavioral therapy, sensory integration occupational therapy, special education, or individualised educational plans, and, when necessary, treatment of physical abnormalities. Persons with fragile X syndrome in their family histories are advised to seek genetic counseling to assess the likelihood of having children who are affected, and how severe any impairments may be in affected descendants.[39]
Medication
Current trends in treating the disorder include medications for symptom-based treatments that aim to minimize the secondary characteristics associated with the disorder. If an individual is diagnosed with FXS, genetic counseling for testing family members at risk for carrying the full mutation or premutation is a critical first-step. Due to a higher prevalence of FXS in boys, the most commonly used medications are stimulants that target hyperactivity, impulsivity, and attentional problems.[7] For co-morbid disorders with FXS, antidepressants such as selective serotonin reuptake inhibitors (SSRIs) are utilized to treat the underlying anxiety, obsessive-compulsive behaviors, and mood disorders. Following antidepressants, antipsychotics such as Risperdal and Seroquel are used to treat high rates of self-injurious, aggressive and aberrant behaviors in this population (Bailey Jr et al., 2012). Anticonvulsants are another set of pharmacological treatments used to control seizures as well as mood swings in 13%–18% of individuals suffering from FXS. Drugs targeting the mGluR5 (metabotropic glutamate receptors) that are linked with synaptic plasticity are especially beneficial for targeted symptoms of FXS.[7] Lithium is also currently being used in clinical trials with humans, showing significant improvements in behavioral functioning, adaptive behavior, and verbal memory. Alongside pharmacological treatments, environmental influences such as home environment and parental abilities as well as behavioral interventions such as speech therapy, sensory integration, etc. all factor in together to promote adaptive functioning for individuals with FXS.[39]
Current pharmacological treatment centers on managing problem behaviors and psychiatric symptoms associated with FXS. However, as there has been very little research done in this specific population, the evidence to support the use of these medications in individuals with FXS is poor.[40]
ADHD, which affects the majority of boys and 30% of girls with FXS, is frequently treated using stimulants.[6] However, the use of stimulants in the fragile X population is associated with a greater frequency of adverse events including increased anxiety, irritability and mood lability.[19] Anxiety, as well as mood and obsessive-compulsive symptoms, may be treated using SSRIs, although these can also aggravate hyperactivity and cause disinhibited behavior.[7][19] Atypical antipsychotics can be used to stabilise mood and control aggression, especially in those with comorbid ASD. However, monitoring is required for metabolic side effects including weight gain and diabetes, as well as movement disorders related to extrapyramidal side effects such as tardive dyskinesia. Individuals with coexisting seizure disorder may require treatment with anticonvulsants.
Prognosis
A 2013 review stated that life expectancy for FXS was 12 years lower than the general population and that the causes of death were similar to those found for the general population.[41]
Research
The increased understanding of the molecular mechanisms of disease in FXS has led to the development of therapies targeting the affected pathways. Evidence from mouse models shows that mGluR5 antagonists (blockers) can rescue dendritic spine abnormalities and seizures, as well as cognitive and behavioral problems, and may show promise in the treatment of FXS.[6][42][43] Two new drugs, AFQ-056 (mavoglurant) and dipraglurant, as well as the repurposed drug fenobam are currently undergoing human trials for the treatment of FXS.[6][44] There is also early evidence for the efficacy of arbaclofen, a GABAB agonist, in improving social withdrawal in individuals with FXS and ASD.[6][13]
In addition, there is evidence from mouse models that minocycline, an antibiotic used for the treatment of acne, rescues abnormalities of the dendrites. An open trial in humans has shown promising results, although there is currently no evidence from controlled trials to support its use.[6]
History
In 1943, James Purdon Martin and Julia Bell described a pedigree of X-linked mental disability, without considering the macroorchidism (larger testicles).[45] In 1969, Herbert Lubs first sighted an unusual "marker X chromosome" in association with mental disability.[46] In 1970, Frederick Hecht coined the term "fragile site". The disorder was also named Escalante syndrome, due to the findings of geneticist Julio Anibal Escalante.
References
- 1 2 3 4 "fragile X syndrome". Genetics Home Reference. April 2012. Retrieved 7 October 2016.
- ↑ Sherman, S. (2002). "Epidemiology". In Hagerman, R. J.; Hagerman, P. J. Fragile X Syndrome, Diagnosis Treatment and Research (3rd ed.). Baltimore: Johns Hopkins University Press. ISBN 0-8018-6843-2.
- 1 2 "What are the treatments for Fragile X syndrome?". www.nichd.nih.gov. Retrieved 2016-11-21.
- ↑ Loomis, E. W.; Eid, J. S.; Peluso, P.; Yin, J.; Hickey, L.; Rank, D.; McCalmon, S.; Hagerman, R. J.; Tassone, F.; Hagerman, P. J. (2012). "Sequencing the unsequenceable: Expanded CGG-repeat alleles of the fragile X gene". Genome Research. 23 (1): 121–8. doi:10.1101/gr.141705.112. PMC 3530672
 . PMID 23064752.
. PMID 23064752. - 1 2 "What are the symptoms of Fragile X syndrome?". www.nichd.nih.gov. Retrieved 2016-11-21.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 McLennan, Y; Polussa J, Tassone F, Hagerman R. (2011). "Fragile X Syndrome". Current Genomics. 12 (3): 216–224. doi:10.2174/138920211795677886. PMC 3137006. PMID 22043169.
- 1 2 3 4 5 6 7 8 9 10 11 Garber, KB; Visootsak J, Warren ST. (2008). "Fragile X syndrome". European Journal of Human Genetics. 16 (6): 666–72. doi:10.1038/ejhg.2008.61. PMID 18398441.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Santoro, MR; Bray SM, Warren ST.; Warren, S. T. (2012). "Molecular Mechanisms of Fragile X Syndrome: A Twenty-Year Perspective". Annu. Rev. Pathol. Mech. Dis. 7: 219–45. doi:10.1146/annurev-pathol-011811-132457. PMID 22017584.
- ↑ Jordan, Joseph A. Regezi, James J. Sciubba, Richard C.K. (2008). "15". Oral pathology : clinical pathologic correlations (5th ed.). St. Louis, Mo.: Saunders/Elsevier. ISBN 978-1-4160-4570-0. Section on Fragile X syndrome
- 1 2 3 Goldstein, Sam; Reynolds, Cecil R. (1999). Handbook of neurodevelopmental and genetic disorders in children. New York: Guilford Press. ISBN 1-57230-448-0.
- 1 2 Hall, Scott S.; Burns, David D.; Lightbody, Amy A.; Reiss, Allan L. (2008). "Longitudinal Changes in Intellectual Development in Children with Fragile X Syndrome". Journal of Abnormal Child Psychology. 36 (6): 927–939. doi:10.1007/s10802-008-9223-y. PMID 18347972.
- 1 2 3 Hagerman, Randi J., and Paul J. Hagerman. Fragile X syndrome: diagnosis, treatment, and research. 3, illustrated ed. Baltimore, MD: JHU P, 2002.
- 1 2 3 4 5 6 7 Budimirovic, DB; Kaufmann WE. (2011). "What can we learn about autism from studying fragile X syndrome?". Dev Neurosci. 33 (5): 379–94. doi:10.1159/000330213. PMC 3254037. PMID 21893949.
- ↑ Pietropaolo S; Guilleminot A; Martin B; D'Amato FR; Crusio WE (2011). "Genetic-background modulation of core and variable autistic-like symptoms in Fmr1 knock-out mice". PLOS ONE. 6 (2): e17073. Bibcode:2011PLoSO...617073P. doi:10.1371/journal.pone.0017073. PMC 3043074. PMID 21364941.
- ↑ Bernardet M; Crusio WE (2006). "Fmr1 KO mice as a possible model of autistic features". The ScientificWorldJournal. 6: 1164–1176. doi:10.1100/tsw.2006.220. PMID 16998604.
- ↑ Mineur, YS; Huynh, LX; Crusio, WE (March 2006). "Social behavior deficits in the Fmr1 mutant mouse". Behavioural Brain Research. 168 (1): 172–175. doi:10.1016/j.bbr.2005.11.004. PMID 16343653.
- ↑ Spencer CM, Alekseyenko O, Hamilton SM, Thomas AM, Serysheva E, Yuva-Paylor LA, Paylor R (February 2011). "Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses". Autism Research. 4 (1): 40–56. doi:10.1002/aur.168. PMC 3059810. PMID 21268289.
- ↑ Spencer CM, Graham DF, Yuva-Paylor LA, Nelson DL, Paylor R (June 2008). "Social behavior in Fmr1 knockout mice carrying a human FMR1 transgene". Behavioral Neuroscience. 122 (3): 710–715. doi:10.1037/0735-7044.122.3.710. PMID 18513141.
- 1 2 3 4 5 6 7 8 9 Tranfaglia, M (2011). "The psychiatric presentation of fragile x: evolution of the diagnosis and treatment of the psychiatric comorbidities of fragile X syndrome". Dev Neurosci. 35 (5): 337–48. doi:10.1159/000329421. PMID 21893938.
- ↑ Holsen, Laura M.; Dalton, Kim M.; Johnstone, Tom; Davidson, Richard J. (2008). "Prefrontal social cognition network dysfunction underlying face encoding and social anxiety in fragile X syndrome". NeuroImage. 43 (3): 592–604. doi:10.1016/j.neuroimage.2008.08.009. PMC 2598775. PMID 18778781.
- ↑ Bourgeois, James A.; Cogswell, Jennifer B.; Hessel, David; Zhang, Lin; Ono, Michele Y.; Tassone, Flora; Farzin, Faraz; Brunberg, James A.; Grigsby, Jim; Hagerman, Randi J. (2007). "Cognitive, anxiety and mood disorders in the fragile X-associated tremor/ataxia syndrome". General Hospital Psychiatry. 29 (4): 349–356. doi:10.1016/j.genhosppsych.2007.03.003. PMC 3991490. PMID 17591512.
- 1 2 3 Berry-Kravis, E; Raspa M, Loggin-Hester L, Bishop E, Holiday D, Bailey Jr DB. (2010). "Seizures in Fragile X Syndrome: Characteristics and Comorbid Diagnoses". Am J Intellect Dev Disabil. 115 (6): 461–72. doi:10.1352/1944-7558-115.6.461. PMID 20945999.
- 1 2 3 4 5 6 7 8 9 Peprah, E (Dec 2011). "Fragile X Syndrome: The FMR1 CGG Repeat Distribution Among World Populations". Annals of Human Genetics. 76 (2): 178–91. doi:10.1111/j.1469-1809.2011.00694.x. PMC 3288311. PMID 22188182.
- ↑ Cornish, Kim; Kogan, Cary S.; Li, Lexin; Turk, Jeremy; Jacquemont, Sebastien; Hagerman, Randi J. (2009). "Lifespan changes in working memory in fragile X premutation males". Brain and Cognition. 69 (3): 551–558. doi:10.1016/j.bandc.2008.11.006. PMID 19114290.
- ↑ Bibi G; Malcov M; Yuval Y; Reches, Adi; Ben-Yosef, Dalit; Almog, Beni; Amit, Ami; Azem, Foad (May 2009). "The effect of CGG repeat number on ovarian response among fragile X premutation carriers undergoing preimplantation genetic diagnosis". Fertil. Steril. 94 (3): 869–74. doi:10.1016/j.fertnstert.2009.04.047. PMID 19481741.
- ↑ Sherman, Stephanie L; Curnow, Eliza C; Easley, Charles A; Jin, Peng; Hukema, Renate K; Tejada, Maria Isabel; Willemsen, Rob; Usdin, Karen (2014). "Use of model systems to understand the etiology of fragile X-associated primary ovarian insufficiency (FXPOI)". Journal of Neurodevelopmental Disorders. 6 (1): 26. doi:10.1186/1866-1955-6-26. ISSN 1866-1947. PMC 4139715. PMID 25147583.
- ↑ Who.int
- 1 2 3 Maddalena, A; Richards C, McGinniss M, Brothman A, Desnick R, Grier R, Hirsch B, Jacky P, McDowell G, Popovich B, Watson M, Wolff D. (2001). "Technical Standards and Guidelines for Fragile X: The First of a Series of Disease-Specific Supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics". Genetics in Medicine. 3 (3): 200–205. doi:10.1097/00125817-200105000-00010. PMC 3110344. PMID 11388762.
- ↑ Nolin SL; Brown WT; Glicksman A; Houck, Jr.; Gargano, Alice D.; Sullivan, Amy; Biancalana, Valérie; Bröndum-Nielsen, Karen; Hjalgrim, Helle; Houck, Elke; Kooy, Frank; Longshore, J; MacPherson, J; Mandel, Jean-Louis; Matthijs, Gert; Rousseau, Francois; Steinbach, Peter; Väisänen, Marja-Leena; von Koskull, Harriet; Sherman, Stephanie L. (2003). "Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles". American Journal of Human Genetics. 72 (2): 454–64. doi:10.1086/367713. PMC 379237. PMID 12529854.
- ↑ Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, Jaffrey SR; Zaninovic, N; Cohen, M. S.; Rosenwaks, Z; Yang, W. Y.; Gerhardt, J; Disney, M. D.; Jaffrey, S. R. (Feb 28, 2014). "Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome". Science. 343 (6174): 1002–5. Bibcode:2014Sci...343.1002C. doi:10.1126/science.1245831. PMID 24578575.
- ↑ Dobyns, William B.; Filauro, Allison; Tomson, Brett N.; Chan, April S.; Ho, Allen W.; Ting, Nicholas T.; Oosterwijk, Jan C.; Ober, Carole (2004). "Inheritance of most X-linked traits is not dominant or recessive, just X-linked". American Journal of Medical Genetics. 129A (2): 136–43. doi:10.1002/ajmg.a.30123. PMID 15316978.
- ↑ Sherman, SL; Jacobs PA, Morton NE, Froster-Iskenius U, Howard-Peebles PN, Nielsen KB, Partington MW, Sutherland GR, Turner G, Watson M. (1985). "Further segregation analysis of the fragile X syndrome with special reference to transmitting males". Hum Genet. 69 (4): 289–99. doi:10.1007/BF00291644. PMID 3838733.
- ↑ Marco, Elysa J.; Skuse, David H. (Dec 2006). "Autism-lessons from the X chromosome". Social Cognitive and Affective Neuroscience. 1 (3): 183–193. doi:10.1093/scan/nsl028. PMC 2555419. PMID 18985105.
- ↑ Bassell GJ, Warren ST; Warren (2008). "Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function". Neuron. 60 (2): 201–14. doi:10.1016/j.neuron.2008.10.004. PMC 3691995. PMID 18957214.
- ↑ Bureau I, Shepherd G, Svoboda K (2008). "Circuit and Plasticity Defects in the Developing Somatosensory Cortex of Fmr1 Knock-Out Mice". The Journal of Neuroscience. 28 (20): 5178–88. doi:10.1523/JNEUROSCI.1076-08.2008. PMID 18480274.
- ↑ Haberl, Matthias G.; Zerbi, Valerio; Veltien, Andor; Ginger, Melanie; Heerschap, Arend; Frick, Andreas (2015-11-01). "Structural-functional connectivity deficits of neocortical circuits in the Fmr1−/y mouse model of autism". Science Advances. 1 (10): e1500775. doi:10.1126/sciadv.1500775. ISSN 2375-2548.
- 1 2 Hogan, A (16 Jan 2012). "Visualizing carrier status: Fragile X sybndrome and genetic diagnosis since the 1940s". Endeavour. 36 (2): 77–84. doi:10.1016/j.endeavour.2011.12.002. PMID 22257912.
- ↑ "How do health care providers diagnose Fragile X syndrome?". www.nichd.nih.gov. Retrieved 2016-11-21.
- 1 2 Hagerman RJ; Berry-Kravis E; Kaufmann WE; Ono, M. Y.; Tartaglia, N.; Lachiewicz, A.; Kronk, R.; Delahunty, C.; Hessl, D.; Visootsak, J.; Picker, J.; et al. (2009). "Advances in the treatment of fragile X syndrome". Pediatrics. 123 (1): 378–90. doi:10.1542/peds.2008-0317. PMC 2888470. PMID 19117905.
- ↑ Rueda JR, Ballesteros J, Tejada MI; Ballesteros; Tejada (2009). "Systematic review of pharmacological treatments in fragile X syndrome". BMC Neurol. 9: 53. doi:10.1186/1471-2377-9-53. PMC 2770029. PMID 19822023.
- ↑ Coppus, A.m.w. (2013-08-01). "People with intellectual disability: What do we know about adulthood and life expectancy?". Developmental Disabilities Research Reviews. 18 (1): 6–16. doi:10.1002/ddrr.1123. ISSN 1940-5529.
- ↑ Dölen G; Osterweil E; Rao BS; Smith, Gordon B.; Auerbach, Benjamin D.; Chattarji, Sumantra; Bear, Mark F. (2007). "Correction of fragile X syndrome in mice". Neuron. 56 (6): 955–62. doi:10.1016/j.neuron.2007.12.001. PMC 2199268. PMID 18093519.
- ↑ Dölen G, Carpenter RL, Ocain TD, Bear MF; Carpenter; Ocain; Bear (2010). "Mechanism-based approaches to treating fragile X". Pharmacol Ther. 127 (1): 78–93. doi:10.1016/j.pharmthera.2010.02.008. PMID 20303363.
- ↑ P. Cole (2012). "Mavoglurant". Drugs of the Future. 37 (1): 7–12. doi:10.1358/dof.2012.037.01.1772147.
- ↑ Martin, J. P. & Bell, J. (1943). "A pedigree of mental defect showing sex-linkage". J. Neurol. Psychiat. 6 (3–4): 154–157. doi:10.1136/jnnp.6.3-4.154. PMC 1090429. PMID 21611430.
- ↑ Lubs, H. A. (1969). "A marker X chromosome". American Journal of Human Genetics. 21 (3): 231–44. PMC 1706424. PMID 5794013.
External links
| Wikimedia Commons has media related to Fragile X syndrome. |
- CDC’s National Center on Birth Defects and Developmental Disabilities
- Fraxa.org – The Fragile X Research Foundation
- Fragilex.org.uk – The United Kingdom National Fragile X charity
- FragileX.org The National Fragile X Foundation (US) – Support, Awareness, Education, Research and Advocacy since 1984
- FragileX.org.au – Fragile X Association of Australia – charity – news, forums, support, information, clinics
- The Colorado Fragile X Consortium
- Gene Reviews