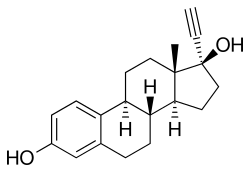
Ethinyl estradiol
 | |
 | |
| Clinical data | |
|---|---|
| Pronunciation | /ˌɛθᵻnᵻlˌiːstrəˈdaɪ.əl/ |
| Trade names | Numerous |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a604032 |
| Pregnancy category |
|
| Routes of administration | Oral, transdermal |
| ATC code | G03CA01 (WHO) L02AA03 (WHO) |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 38–48%[1][2] |
| Protein binding | 97–98% (to albumin;[3] does not bind to SHBG)[4] |
| Metabolism | Liver (CYP3A4)[5] |
| Biological half-life | 7–36 hours[6][1][5][7] |
| Excretion |
Feces: 62%[6] Urine: 38%[6] |
| Identifiers | |
| |
| Synonyms | (17α)-19-Norpregna-1,3,5(10)-trien-20-yne-3,17β-diol |
| CAS Number |
57-63-6 |
| PubChem (CID) | 5991 |
| IUPHAR/BPS | 7071 |
| DrugBank |
DB00977 |
| ChemSpider |
5770 |
| UNII |
423D2T571U |
| KEGG |
D00554 |
| ChEBI |
CHEBI:4903 |
| ChEMBL |
CHEMBL691 |
| Chemical and physical data | |
| Formula | C20H24O2 |
| Molar mass | 296.403 g/mol |
| 3D model (Jmol) | Interactive image |
| |
| |
| (verify) | |
Ethinyl estradiol (EE2) (USP) (former brand name Estinyl), or ethinylestradiol (INN, USAN, JAN), also spelled as ethinylœstradiol (BAN) and also known as 17α-ethynylestra-1,3,5(10)-triene-3,17β-diol, is a synthetic, steroidal estrogen.[8][9][10] It is a derivative of estradiol, the major endogenous estrogen in humans; specifically, EE2 is 17α-ethynylestradiol.[8] EE2 is an orally active estrogen used in almost all formulations of combined oral contraceptives (COCs), being nearly the exclusive estrogen used for this purpose.[11]
Medical uses
As Estinyl, EE2 was formerly used for hormone replacement therapy in menopause and the treatment of female hypogonadism, loss of menstruation, dysmenorrhea, acne, prostate cancer, and breast cancer.[12] However, in more recent times, EE2 is mainly used in COCs. In contraception, due to concerns of unopposed estrogen action and the possible increased risk of endometrial cancer that accompanies this, EE2 is formulated in combination with progestins. EE2 is little used in menopausal hormone replacement therapy.[13]
EE2 has been used at very high dosages (1–2 mg/day) in the treatment of prostate cancer.[14]
Contraindications
EE2 should be avoided in women with a history of or known susceptibility to thrombosis (blood clots), particularly venous thromboembolism (VTE).
Due to risk of cholestatic hepatotoxicity, it is widely considered that COCs containing EE2 should be avoided in women with a history of cholestasis of pregnancy, hepatic tumors, active hepatitis, and familial defects in biliary excretion.[15]
Side effects
Side effects of EE2 are the same as for other estrogens and include breast tenderness, headache, fluid retention (bloating), nausea, dizziness, and weight gain.[6][15] The estrogen component of oral contraceptives, which is almost always EE2, can cause breast tenderness and fullness.[16]
Rare reactions
Venous thromboembolism
EE2 carries a greater risk of blood clot formation and VTE than does natural estradiol, which is thought to be due to different degrees of hepatic metabolism between the two drugs (see below).
The original formulations of COCs contained as much as 150 μg EE2.[17] However, it was soon found that EE2 is associated with incidence of VTE and that the risk is dose-dependent.[17] Subsequently, the dosage of EE2 was greatly reduced, and is now generally between 25 and 35 μg,[17] in some cases less than 20 μg,[17] and not more than 50 μg.[18][19][16] These lower dosages have a significantly reduced risk of VTE with no loss of contraceptive effectiveness.[17] According to a bulletin posted by the U.S. FDA, the rate of deep vein thrombosis in women taking COCs containing 20 to 40 μg EE2 is 4 per 10,000, which is approximately equivalent to the rate of 3 per 10,000 in women not taking a COC.[20] No study has shown a further reduced risk of VTE below an EE2 dosage of 30 or 35 μg.[17]
Cholestatic hepatotoxicity
EE2 has, albeit rarely (at the low dosages that are now used in COCs), been associated with cholestatic hepatotoxicity similarly to 17α-alkylated anabolic-androgenic steroids and 17α-ethynylated 19-nortestosterone progestins.[21][22] Glucuronide metabolites of EE2, via effects on the ABCB11 (BSEP) and MRP2 (ABCC2) proteins and consequent changes in bile flow and bile salt excretion, appear to be responsible for the cholestasis.[23] High concentrations of estradiol, via its metabolite estradiol D-glucuronide, are also implicated in cholestasis, for instance in cholestasis of pregnancy.[22] However, the incidence and severity of cholestatic hepatotoxicity appear to be much greater with EE2 than with estradiol, which is due to its 17α-ethynyl substitution and consequent reduced metabolism.[15]
Endometrial cancer
The high doses of EE2 that were used in early COCs were associated with a significantly increased risk of endometrial cancer in certain preparations, for instance those containing the progestogen dimethisterone.[24] Unopposed estrogens like EE2 have carcinogenic effects in the endometrium and progestogens protect against these effects, but dimethisterone is a relatively weak progestogen and was unable to adequately antagonize the endometrial carcinogenic effects of EE2, in turn resulting in the increased risk of endometrial cancer.[24] COCs containing dimethisterone have since been discontinued (with more potent progestogens used instead) and doses of EE2 in COCs in general have been dramatically reduced, abrogating the risk.[24] In turn, most studies of modern COCs have found a decreased risk of endometrial cancer.[25]
Interactions
Inducers of certain cytochrome P450 enzymes such as CYP3A4 can decrease circulating concentrations of EE2.[15] Examples include anticonvulsants like phenytoin, primidone, ethosuximide, phenobarbital, and carbamazepine, azole antifungals like fluconazole, and rifamycin antibiotics like rifampin (rifampicin).[15] Conversely, inhibitors of CYP3A4 and certain other cytochrome P450 enzymes may increase circulating levels of EE2.[15] An example is troleandomycin, which is a potent and highly selective inhibitor of CYP3A4.[15]
Paracetamol has been found to competitively inhibit the sulfation of EE2, with pretreatment of 1 g paracetamol significantly increasing the AUC levels of EE2 (by 22%) and decreasing the AUC levels of EE2 sulfate in women.[15] The same has been found for ascorbic acid (vitamin C) and EE2, although the significance of the interaction has been regarded as dubious.[15]
Unlike the case of estradiol, there is probably no pharmacokinetic interaction between smoking (which potently induces certain cytochrome P450 enzymes and markedly increases the 2-hydroxylation of estradiol) and EE2.[15] This suggests that estradiol and EE2 are metabolized by different cytochrome P450 enzymes.[15] There is however still an increased risk of cardiovascular complications with smoking and EE2, similarly to the case of smoking and other estrogens.[15]
The 19-nortestosterone progestins, gestodene and, to a lesser extent, desogestrel, have been found to inhibit cytochrome P450 enzymes and to progressively inhibit the metabolism and increase the concentrations of EE2.[15]
EE2 has been found to significantly increase (by 38%) the AUC of omeprazole (which is metabolized by CYP2C19).[15]
Pharmacology
EE2 is an estrogen similarly to natural estrogens like estradiol and conjugated equine estrogens and synthetic estrogens like diethylstilbestrol. It binds to and activates both isoforms of the estrogen receptor, ERα and ERβ.[13] In one study, EE2 was found to have 233% and 37.8% of the affinity of estradiol for the ERα and ERβ, respectively.[26] EE2 also appears to signal through the GPER membrane estrogen receptor, similarly to estradiol.[27][28][29]
Orally, EE2 is about 100 times as potent by weight as natural estrogens like micronized estradiol and conjugated equine estrogens.[30][31][32] In contrast, the potencies of EE2 and natural estrogens are similar when they are administered intravenously, due to the bypassing of first-pass metabolism.[17] Relative to its prodrug mestranol, EE2 is about 1.7 times as potent by weight orally.[31]
As can be seen in the table below, EE2 shows strong and disproportionate effects on hepatic protein production relative to estradiol.[33] The liver as well as the uterus express 17β-hydroxysteroid dehydrogenase (17β-HSD), and this enzyme serves to inactivate estradiol and effectively suppress its potency in these tissues (analogously but in the opposite manner to potentiation of testosterone by 5α-reductase into the more potent dihydrotestosterone in so-called androgenic tissues like the skin, hair follicles, and prostate gland)[34] by reversibly converting it into the far less potent estrogen estrone (which has approximately 4% of the estrogenic activity of estradiol, most of which is actually due to conversion into estradiol).[33] In contrast to estradiol, the 17α-ethynyl group of EE2 prevents oxidation of the C17β position of EE2 by 17β-HSD, and for this reason, EE2 is not inactivated in these tissues and has much stronger relative estrogenic activity in them.[33][35][7] This is the mechanism of the disproportionately strong effects of EE2 on hepatic protein production,[33][35] which results in a greatly increased magnitude of effect on VTE risk relative to estradiol.[36]
On the other hand, due to the loss of inactivation of EE2 by 17β-HSD in the endometrium (uterus), EE2 is relatively more active than estradiol in the endometrium and, for this reason, is associated with a significantly lower incidence of vaginal bleeding and spotting in comparison.[33] This is particularly so in the case of combined estrogen and progestogen therapy (as in COCs or menopausal HRT), as progestogens induce the expression of 17β-HSD in the endometrium.[33] The reduced vaginal bleeding and spotting with EE2 is one of the main reasons that it is used in COCs instead of estradiol,[2] in spite of its potentially inferior safety profile (related to its adverse effects on hepatic protein synthesis and VTE incidence).[37]
EE2 has been found to have similar effects on hepatic protein production and VTE risk regardless of whether the route of administration is oral, transdermal, or vaginal, indicating that oral versus non-oral routes do not reduce the hepatic actions of EE2 relative to non-hepatic actions.[35] In contrast, at typical menopausal dosages, whereas oral estradiol shows significant effects on hepatic protein production, transdermal estradiol shows little or no such effects.[33]
| Estrogen | Hot flashes | FSH | HDL cholesterol | SHBG | CBG | Angiotensinogen | |||
|---|---|---|---|---|---|---|---|---|---|
| Estradiol | 100 | 100 | 100 | 100 | 100 | 100 | |||
| Estriol | 30 | 30 | 20 | ? | ? | ? | |||
| Estrone sulfate | ? | 90 | 50 | 90 | 70 | 150 | |||
| CEEs | 120 | 110 | 150 | 300 | 150 | 500 | |||
| Equilin sulfate | ? | ? | 600 | 750 | 600 | 750 | |||
| Ethinyl estradiol | 12,000 | 12,000 | 40,000 | 50,000 | 60,000 | 35,000 | |||
| Diethylstilbestrol | ? | 340 | ? | 2,560 | 2,450 | 1,950 | |||
| Hot flashes = clinical relief of hot flashes; FSH = suppression of FSH levels; HDL cholesterol, SHBG, CBG, and angiotensinogen = increase in the serum levels of these hepatic proteins. | |||||||||
Pharmacokinetics
Absorption
The oral bioavailability of EE2 is between 38 to 48%, with a wide range of 20% to 65% (mean 45%) that is due to high interindividual variability.[6] Although relatively low, the oral bioavailability of EE2 is considerably higher than that of estradiol (5%).[1][6] Following a single 20 μg dose of EE2 in combination with 1 mg norethisterone in postmenopausal women, EE2 concentrations have been found to reach a maximum of 50 pg/mL within an average of 1.5 hours.[33] Following the first dose, mean levels of EE2 in general further increase by about 50% until steady-state concentrations are reached;[33] steady-state is reached after one week of daily administration.[13] For comparison, the mean peak levels of estradiol achieved with 2 mg micronized estradiol or estradiol valerate are 40 pg/mL following the first dose and 80 pg/mL after three weeks of administration.[33] These concentrations of estradiol are in the same range as the concentrations of EE2 that are produced by an oral dose of EE2 that is 100 times lower by weight, which is in accordance with the approximately 100-fold increased oral potency of EE relative to estradiol.[30][33] In accordance with the high interindividual variability in the oral bioavailability of EE2, there is a large degree of interindividual variation in EE2 levels.[33]
Distribution
Unlike estradiol, which binds with high affinity to sex hormone-binding globulin (SHBG), EE2 has no affinity for this protein and is instead bound almost exclusively to albumin (97–98%).[3][33][6][38] As estradiol that is bound to SHBG is considered to be hormonally inactive,[39] the lack of binding of EE2 to SHBG may be involved in its increased comparative potency.
Metabolism
Due to high first-pass metabolism in the intestines and liver, only 1% of an oral dose of an EE2 appears in the circulation as EE2 itself.[33] During first-pass metabolism, EE is extensively conjugated via sulfation into the hormonally inert EE2 sulfate, and levels of EE2 sulfate in circulation are between 6- and 22-fold higher than those of EE2.[33] For comparison, with oral administration of 2 mg micronized estradiol, levels of estrone and estrone sulfate are 4- to 6-fold and 200-fold higher than those of estradiol, respectively.[33] In contrast to estradiol, EE2, due to steric hindrance by its 17α-ethynyl group, is not metabolized or inactivated by 17β-HSD,[7] and this is the primary factor responsible for the dramatically increased potency of oral EE2 relative to oral estradiol.[33] Due to the formation of EE2 sulfate, enterohepatic circulation is involved in the pharmacokinetics of EE2 similarly to estradiol, although to a lesser extent.[33]
Aside from sulfate conjugation, EE2 is mainly metabolized by hydroxylation into catechol estrogens.[33] This is mainly by 2-hydroxylation into 2-hydroxy-EE2, which is catalyzed primarily by CYP3A4.[6] Hydroxylation of EE2 at the C4, C6α, and C16β positions into 4-, 6α-, and 16β-hydroxy-EE2 has also been reported, but appears to contribute to its metabolism to only a small extent.[6] 2- and 4-methoxy-EE2 are also formed via transformation by catechol O-methyltransferase of 2- and 4-hydroxy-EE.[33] Unlike the case of estradiol, 16α-hydroxylation does not occur with EE2, owing to steric hindrance by its ethynyl group at C17α.[6][33] The ethynylation of EE2 is largely irreversible, and so EE2 is not metabolized into estradiol, unlike estradiol esters.[33] A review found that the range of the reported terminal half-life of EE in the literature was 13.1 to 27.0 hours.[1] Another review reported a terminal half-life of EE2 of 10–20 hours.[6] However, the terminal half-life of EE2 has also been reported by other sources to be as short as 7 hours[7] and as long as 36 hours[5]
Unlike the case of estradiol, in which there is a rapid rise in its levels and which remain elevated in a plateau-like curve for many hours, levels of EE2 fall rapidly after peaking.[33] This is thought to be because estrone and estrone sulfate can be reversibly converted back into estradiol and serve as a hormonally inert reservoir for estradiol, whereas the EE2 sulfate reservoir for EE2 is much smaller in comparison.[33]
EE2, following oxidative formation of a very reactive metabolite, irreversibly inhibits cytochrome P450 enzymes involved in its metabolism, and this may also role in the increased potency of EE2 relative to estradiol.[33] Indeed, EE2 is said to have a marked effect on hepatic metabolism, and this is one of the reasons, among others, that natural estrogens like estradiol may be preferable.[38]
Elimination
EE2 is eliminated 62% in the feces and 38% in the urine.[6]
Chemistry
EE2 is an estrane (C18) steroid and derivative of estradiol with an ethynyl substitution at the C17α position. It is also known as 17α-ethynylestradiol or as 17α-ethynylestra-1,3,5(10)-triene-3,17β-diol. The 17α-ethynylation of EE2 is analogous to the 17α-substitution of testosterone derivatives such as 17α-ethynylated progestins like ethisterone and norethisterone and 17α-alkylated anabolic-androgenic steroids like methyltestosterone.
Derivatives
A number of derivatives of EE exist. These include mestranol (EE 3-methyl ether), quinestrol (EE 3-cyclopentyl ether), ethinyl estradiol sulfonate (EE 3-isopropylsulfonate), and moxestrol (11β-methoxy-EE). The former three are prodrugs of EE, while the latter is not.
Analogues
A few 17α-substituted analogues of EE exist. Examples include methylestradiol, ethinyl estriol, and nilestriol (ethinyl estriol 3-cyclopentyl ether).
History
The first orally active semisynthetic steroidal estrogen, EE2 (17α-ethynylestradiol), the 17α-ethynyl analogue of estradiol, was synthesized in 1938 by Hans Herloff Inhoffen and Walter Hohlweg at Schering AG in Berlin.[40][41][42][43][44] EE2 was approved by the FDA in the U.S. on June 25, 1943 and marketed by Schering as Estinyl.[45] The FDA withdrew approval of Estinyl effective June 4, 2004 at the request of Schering, which had discontinued marketing it.[46] EE2 has been marketed as a standalone drug under the brand names Estinyl, Feminone, Lynoral, Novestrol, and Palonyl, although most or all of these formulations are now discontinued.[47][10]
EE2 was first used in COCs, as an alternative to mestranol, in 1964, and eventually superseded mestranol in COCs.[48]
References
- 1 2 3 4 Goldzieher JW, Brody SA (1990). "Pharmacokinetics of ethinyl estradiol and mestranol". American Journal of Obstetrics and Gynecology. 163 (6 Pt 2): 2114–9. PMID 2256522.
- 1 2 Fruzzetti F, Trémollieres F, Bitzer J (2012). "An overview of the development of combined oral contraceptives containing estradiol: focus on estradiol valerate/dienogest". Gynecological Endocrinology : the Official Journal of the International Society of Gynecological Endocrinology. 28 (5): 400–8. doi:10.3109/09513590.2012.662547. PMC 3399636
 . PMID 22468839.
. PMID 22468839. - 1 2 Facts and Comparisons (Firm); Ovid Technologies, Inc (2005). Drug Facts and Comparisons 2005: Pocket Version. Facts and Comparisons. p. 121. ISBN 978-1-57439-179-4.
- ↑ Micromedex (1 January 2003). USP DI 2003: Drug Information for Healthcare Professionals. Thomson Micromedex. pp. 1253,1258,1266. ISBN 978-1-56363-429-1.
- 1 2 3 Claude L Hughes; Michael D. Waters (23 March 2016). Translational Toxicology: Defining a New Therapeutic Discipline. Humana Press. pp. 73–. ISBN 978-3-319-27449-2.
- 1 2 3 4 5 6 7 8 9 10 11 12 Stanczyk FZ, Archer DF, Bhavnani BR (2013). "Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment". Contraception. 87 (6): 706–27. doi:10.1016/j.contraception.2012.12.011. PMID 23375353.
- 1 2 3 4 Shellenberger, T. E. (1986). "Pharmacology of estrogens": 393–410. doi:10.1007/978-94-009-4145-8_36.
Ethinyl estradiol is a synthetic and comparatively potent estrogen. As a result of the alkylation in 17-C position it is not a substrate for 17(3- dehydrogenase, an enzyme which transforms natural estradiol-17 (3 to the less potent estrone in target organs.
- 1 2 J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 522–. ISBN 978-1-4757-2085-3.
- ↑ I.K. Morton; Judith M. Hall (6 December 2012). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 115–. ISBN 978-94-011-4439-1.
- 1 2 Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. p. 412. ISBN 978-3-88763-075-1.
- ↑ Evans G, Sutton EL. Oral contraception. Med Clin North Am. 2015 May;99(3):479-503. PMID 25841596
- ↑ RxList.com - Estinyl (ethynyl estradiol)
- 1 2 3 Michael Oettel; Ekkehard Schillinger (6 December 2012). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen. Springer Science & Business Media. pp. 248,369. ISBN 978-3-642-60107-1.
The binding affinity of EE2 for the estrogen receptor is similar to that of estradiol. [...] During daily intake, the EE2 levels increase up to a steady state which is reached after about 1 week.
- ↑ R. S. Satoskar; S. D. Bhandarkar &nirmala N. Rege (1973). Pharmacology and Pharmacotherapeutics. Popular Prakashan. pp. 934–. ISBN 978-81-7991-527-1.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Jeffrey K. Aronson (21 February 2009). Meyler's Side Effects of Endocrine and Metabolic Drugs. Elsevier. pp. 177,219,223,224,230,232,239,242. ISBN 978-0-08-093292-7.
- 1 2 Kenneth L. Becker (2001). Principles and Practice of Endocrinology and Metabolism. Lippincott Williams & Wilkins. pp. 1024,1035. ISBN 978-0-7817-1750-2.
Low-dose COCs contain <50 μg of estrogen and are the primary choice for oral contraception. COCs containing ≥50 μg of estrogen should no longer be routinely used for contraception. [...] The estrogen component of COCs can cause breast fullness and tenderness.
- 1 2 3 4 5 6 7 Tommaso Falcone; William W. Hurd (2007). Clinical Reproductive Medicine and Surgery. Elsevier Health Sciences. pp. 388–. ISBN 0-323-03309-1.
- ↑ Committee on the Relationship Between Oral Contraceptives and BreastCancer (1 January 1991). Oral Contraceptives and Breast Cancer. National Academies. pp. 143–. NAP:13774.
Following a recommendation by its Fertility and Maternal Health Drugs Advisory Committee, the Food and Drug Administration (FDA) recently ordered the removal from the market of all oral contraceptives with [ethinyl estradiol] contents greater than 50 μg.
- ↑ Multigenerational Reproductive Toxicology Study of Ethinyl Estradiol (CAS No. 57636) in SpragueDawley Rats (Feed Studies). DIANE Publishing. pp. 27–. ISBN 978-1-4379-4231-6.
Oral contraceptive formulations containing greater than 50 ug ethinyl estradiol were removed from the United States market in 1989, and currently marketed formulations generally contain between 20 and 35 μg ethinyl estradiol.
- ↑ Daniel Jeffrey Wallace; Bevra Hahn (2007). Dubois' Lupus Erythematosus. Lippincott Williams & Wilkins. pp. 1252–. ISBN 978-0-7817-9394-0.
- ↑ Michael Trauner; Peter L. M. Jansen (2004). Molecular Pathogenesis of Cholestasis. Springer Science & Business Media. pp. 260–. ISBN 978-0-306-48240-3.
- 1 2 Pierre-Alain Clavien; John Baillie (15 April 2008). Diseases of the Gallbladder and Bile Ducts: Diagnosis and Treatment. John Wiley & Sons. pp. 363–. ISBN 978-0-470-98697-4.
- ↑ Peter J. O'Brien; William Robert Bruce (2010). Endogenous Toxins: Diet, Genetics, Disease and Treatment. John Wiley & Sons. pp. 302–. ISBN 978-3-527-32363-0.
- 1 2 3 A. Blaustein (11 November 2013). Pathology of the Female Genital Tract. Springer Science & Business Media. pp. 291–. ISBN 978-1-4757-1767-9.
- ↑ Earl A. Surwit; David Alberts (6 December 2012). Endometrial Cancer. Springer Science & Business Media. pp. 11–. ISBN 978-1-4613-0867-6.
- ↑ Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V, Balaguer P (2006). "Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta". Biochem. Pharmacol. 71 (10): 1459–69. doi:10.1016/j.bcp.2006.02.002. PMID 16554039.
- ↑ Yates MA, Li Y, Chlebeck PJ, Offner H (2010). "GPR30, but not estrogen receptor-alpha, is crucial in the treatment of experimental autoimmune encephalomyelitis by oral ethinyl estradiol". BMC Immunol. 11: 20. doi:10.1186/1471-2172-11-20. PMC 2864220. PMID 20403194.
- ↑ Prossnitz ER, Barton M (2011). "The G-protein-coupled estrogen receptor GPER in health and disease". Nat Rev Endocrinol. 7 (12): 715–26. doi:10.1038/nrendo.2011.122. PMC 3474542. PMID 21844907.
Further research showed that the therapeutic effect of ethynylestradiol in established EAE was mediated via GPER, but not via ERα, and possibly involved production of the anti-inflammatory cytokine Il‑10.115
- ↑ Prossnitz ER, Barton M (2014). "Estrogen biology: new insights into GPER function and clinical opportunities". Mol. Cell. Endocrinol. 389 (1-2): 71–83. doi:10.1016/j.mce.2014.02.002. PMC 4040308. PMID 24530924.
In addition, the therapeutic effect of ethinyl estradiol in established disease was demonstrated to require expression of GPER but not ERα, and was associated with the production of the anti-inflammatory cytokine IL-10 (Yates et al., 2010).
- 1 2 Victor Gomel; Malcolm G. Munro; Timothy C. Rowe (1990). Gynecology: a practical approach. Williams & Wilkins. p. 132,134. ISBN 978-0-683-03631-2.
The synthetic estrogen, ethinyl estradiol, more commonly used in oral contraceptives, has a biological activity 100 times that of the native and conjugated substances.
- 1 2 Donna Shoupe (7 November 2007). The Handbook of Contraception: A Guide for Practical Management. Springer Science & Business Media. pp. 23–. ISBN 978-1-59745-150-5.
EE2 has about 100 times the potency of an equivalent weight of conjugated equine estrogen or estrone sulfate for stimulating synthesis of hepatic proteins. [...] EE2 is about 1.7 times as potent as the same weight of mestranol.
- ↑ Nathaniel McConaghy (21 November 2013). Sexual Behavior: Problems and Management. Springer Science & Business Media. pp. 177–. ISBN 978-1-4899-1133-9.
Meyer et al. found that ethinyl estradiol was 75 to 100 times more potent than conjugated estrogen on the basis of the doses required to lower testosterone to the adult female range, 0.1 mg of the former and 7.5 to 10 mg of the latter being necessary.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 Kuhl H (2005). "Pharmacology of estrogens and progestogens: influence of different routes of administration". Climacteric. 8 Suppl 1: 3–63. doi:10.1080/13697130500148875. PMID 16112947.
- ↑ Jerome F. Strauss, III; Robert L. Barbieri (13 September 2013). Yen and Jaffe's Reproductive Endocrinology. Elsevier Health Sciences. pp. 83–. ISBN 978-1-4557-2758-2.
- 1 2 3 Rogerio A. Lobo (5 June 2007). Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. Academic Press. pp. 177,770–771. ISBN 978-0-08-055309-2.
- ↑ Donna Shoupe (10 February 2011). Contraception. John Wiley & Sons. pp. 79–. ISBN 978-1-4443-4263-5.
- ↑ Sitruk-Ware R, Nath A (2011). "Metabolic effects of contraceptive steroids". Rev Endocr Metab Disord. 12 (2): 63–75. doi:10.1007/s11154-011-9182-4. PMID 21538049.
- 1 2 M. Notelovitz; P.A. van Keep (6 December 2012). The Climacteric in Perspective: Proceedings of the Fourth International Congress on the Menopause, held at Lake Buena Vista, Florida, October 28–November 2, 1984. Springer Science & Business Media. pp. 395–. ISBN 978-94-009-4145-8.
- ↑ Leonid Poretsky (24 January 2010). Principles of Diabetes Mellitus. Springer Science & Business Media. pp. 591–. ISBN 978-0-387-09841-8.
- ↑ Inhoffen, H. H.; Hohlweg, W. (1938). "Neue per os-wirksame weibliche Keimdrüsenhormon-Derivate: 17-Aethinyl-oestradiol und Pregnen-in-on-3-ol-17 (New female glandular derivatives active per os: 17α-ethynyl-estradiol and pregnen-in-on-3-ol-17)". Naturwissenschaften. 26 (6): 96. doi:10.1007/BF01681040.
- ↑ Maisel, Albert Q. (1965). The Hormone Quest. New York: Random House. OCLC 543168.
- ↑ Petrow, Vladimir (December 1970). "The contraceptive progestagens". Chem Rev. 70 (6): 713–26. doi:10.1021/cr60268a004. PMID 4098492.
- ↑ Sneader, Walter (2005). "Hormone analogues". Drug discovery : a history. Hoboken, NJ: John Wiley & Sons. pp. 188–225. ISBN 0-471-89980-1.
- ↑ Djerassi, Carl (January 2006). "Chemical birth of the pill". American Journal of Obstetrics and Gynecology. 194 (1): 290–8. doi:10.1016/j.ajog.2005.06.010. PMID 16389046.
- ↑ FDA (2007). "Approval history: Estinyl (ethinyl estradiol) NDA 005292". search: Estinyl
- ↑ FDA (May 5, 2004). "Schering Corp. et al.; Withdrawal of Approval of 92 New Drug Applications and 49 Abbreviated New Drug Applications. Notice" (PDF). Federal Register. 69 (87): 25124–30.
- ↑ Council on Drugs (American Medical Association) (1977). AMA Drug Evaluations. American Medical Association.
Ethinyl Estradiol [Estinyl, Feminone, Lynoral, Novestrol, Palonyl]
- ↑ J.G. Gruhn; R.R. Kazer (11 November 2013). Hormonal Regulation of the Menstrual Cycle: The Evolution of Concepts. Springer Science & Business Media. pp. 185–. ISBN 978-1-4899-3496-3.
In 1964, ethinyl estradiol was introduced as an alternative to mestranol as the estrogenic component, [...]