Nav1.8
| View/Edit Human | View/Edit Mouse |
Nav1.8 is a sodium ion channel that in humans is encoded by the SCN10A gene.[3] Nav1.8 is a sodium channel subunit.[4][5][6]
Nav1.8 is a tetrodotoxin (TTX)-resistant voltage-gated sodium ion channel. It is expressed specifically in the dorsal root ganglion (DRG), in unmyelinated, small-diameter sensory neurons called C-fibres and is involved in the pain pathway called nociception.[7][8] C-fibres can be activated by noxious thermal or mechanical stimuli and thus can carry pain messages.
The discrete locations of Nav1.8 in sensory neurons of the DRG may be the key factor in therapeutic targets for the development of new analgesics[9] and to treat chronic pain.[10]
Function
Voltage-gated sodium ion channels (VGSC) are essential in producing and propagating action potentials. Tetrodotoxin, a toxin found in pufferfish is able to block some VGSCs and therefore is used to distinguish the different subtypes. There are three TTX-resistant VGSC: Nav1.5, Nav1.8 and Nav1.9. Nav1.8 and Nav1.9 are both expressed in nociceptors (damage-sensing neurons). It is known that Nav1.7, Nav1.8 and Nav1.9 are located in the DRG and play an important role in maintaining chronic inflammatory pain.[11]
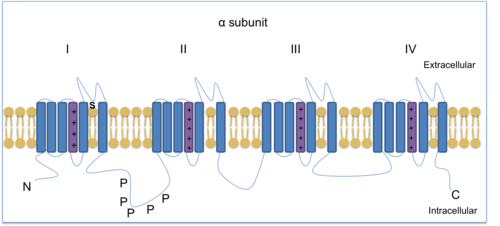
Nav1.8 α-subunit consists of four homologous domains each with six transmembrane spanning regions of which one is a voltage sensor. Voltage clamp methods were used to show how action potentials in small diameter DRG cell bodies are shaped by TTX-resistant sodium channels. Nav1.8 contributes the most to sustaining the depolarising stage of the action potentials in nociceptive sensory neurons by activating quickly and remaining activated[12][13] after detecting a noxious stimulus.
Therefore, Nav1.8 is known to perform a key role in hyperalgesia (increased sensitivity to pain) and allodynia (pain from stimuli that do not usually cause it) which are part of chronic pain.[14] Nav1.8 knockout mice studies have shown that the channel is associated with inflammatory and neuropathic pain.[7][15][16] Moreover, Nav1.8 plays a crucial role in cold pain.[17] Zimmermann et al. show that reducing the temperature from 30 °C to 10 °C slows the activation of VGSCs and hence decreases the current. However, Nav1.8 is cold-resistant and is able to generate action potentials in the cold to carry information from nociceptors to the central nervous system (CNS). Furthermore, Nav1.8-null mice failed to produce action potentials therefore indicating Nav1.8 is essential in the notion of pain in cold temperatures.[17]
Clinical significance
Pain signalling pathways
Nociceptors are different from other sensory neurons in that they have a low activating threshold and consequently increase their response to constant stimuli. Therefore, nociceptors are easily sensitised by agents such as bradykinin and nerve growth factor (NGF), which are released at the site of tissue injury, ultimately causing changes to ion channel conductance. VGSCs have been shown to increase in density after nerve injury.[18] Therefore, VGSCs can be modulated by many different hyperalgesic agents that are released after nerve injury and further examples include prostaglandin E2 (PGE2), serotonin (5-HT) and adenosine, which all act to increase the current through Nav1.8.[19]
Prostaglandins (e.g. PGE2) can sensitise nociceptors to thermal, chemical and mechanical stimuli and increase the excitability of DRG sensory neurons. This occurs due to PGE2 modulating the trafficking of Nav1.8 by binding to G-protein coupled EP2 receptor which in turn activates protein kinase A (PKA).[20] PGE2 modulates the trafficking of Nav1.8 by binding to G-protein coupled EP2 receptor which in turn activates protein kinase A (PKA).[21] PKA phosphorylates Nav1.8 at intracellular sites resulting in increased sodium ion currents. Evidence for PGE2 linking to hyperalgesia comes from an antisense deoxynucleotide knockdown of Nav1.8 in the DRG of rats.[22] Another modulator of Nav1.8 is the ε isoform of PKC (PKCε). PKCε is activated by inflammatory mediator bradykinin and phosphorylates Nav1.8 causing an increase in sodium current in the sensory neurons, which plays an important role is mechanical hyperalgesia.[23]
Brugada Syndrome
Mutations in SCN10A are associated to Brugada Syndrome .[24]
Membrane trafficking
Nerve growth factor (NGF) levels in inflamed or injured tissues are increased creating an increased sensitivity to pain (hyperalgesia).[25] The increased levels of NGF and tumour necrosis factor-α (TNF-α) causes the upregulation of Nav1.8 in sensory neurons via the accessory protein p11 (annexin II light chain). It has been shown using the yeast-two hybrid screening method that p11 binds to a 28 amino acid fragment of the N-terminal of Nav1.8 and promotes its translocation to the plasma membrane. This contributes to the hyperexcitability of sensory neurons during pain.[26] p11-null nociceptive sensory neurons in mice, created using the Cre-LoxP recombinase system, showed a decrease in Nav1.8 expression at the plasma membrane.[27] Therefore, disrupting the interactions between p11 and Nav1.8 may be a good therapeutic target to lower pain.
In myelinated fibres VGSCs are located at the nodes of Ranvier however, in unmyelinated fibres the exact location of VGSCs have not been found. Nav1.8 is located in unmyelinated fibres and has been found to be located in clusters associated with lipid rafts along DRG neurons both in vitro and in vivo.[28] Lipid rafts organise the cell membrane, which includes trafficking and localising ion channels. Removal of lipid rafts in the membrane using MβCD, which depletes cholesterol in the plasma membrane that is crucial for lipid rafts, leads to a shift of Nav1.8 to a non-raft portion of the membrane, causing reduced action potential firing and propagation.[28]
Painful peripheral neuropathies
Painful peripheral neuropathies or small-fibre neuropathies are disorders of unmyelinated nociceptive C-fibres causing neuropathic pain and in some cases there is no known cause.[29] Patients with these idiopathic neuropathies have been screened genetically to find mutations in the SCN9A gene encoding Nav1.7. A gain-of-function mutation in Nav1.7 located in the DRG sensory neurons was found in 30% of patients.[30] This gain-of-function mutation causes an increase in excitability (hyperexcitability) of DRG sensory neurons thus causing an increase in pain. Nav1.7 has therefore been shown to be linked to human pain however, Nav1.8 had only been associated to pain in animal studies until recently. A gain-of-function mutation was found in the Nav1.8 encoding SCN10A gene in patients with painful peripheral neuropathy.[31] Faber et al. used voltage clamp and current clamp methods along with predictive algorithms on 104 idiopathic patients who did not have the mutation in SCN9A. They found two gain-of-function mutations in SCN10A in three patients in which both mutations cause increased excitability in DRG sensory neurons and hence they contribute to pain but the mechanism in which they do so is not understood.
References
- ↑ "Human PubMed Reference:".
- ↑ "Mouse PubMed Reference:".
- ↑ "Entrez Gene: sodium channel".
- ↑ Rabert DK, Koch BD, Ilnicka M, Obernolte RA, Naylor SL, Herman RC, Eglen RM, Hunter JC, Sangameswaran L (November 1998). "A tetrodotoxin-resistant voltage-gated sodium channel from human dorsal root ganglia, hPN3/SCN10A". Pain. 78 (2): 107–14. doi:10.1016/S0304-3959(98)00120-1. PMID 9839820.
- ↑ Plummer NW, Meisler MH (April 1999). "Evolution and diversity of mammalian sodium channel genes". Genomics. 57 (2): 323–31. doi:10.1006/geno.1998.5735. PMID 10198179.
- ↑ Catterall WA, Goldin AL, Waxman SG (December 2005). "International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels". Pharmacol. Rev. 57 (4): 397–409. doi:10.1124/pr.57.4.4. PMID 16382098.
- 1 2 Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH & Wood JN (1999). "The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways". Nature Neuroscience. 2: 541–548. doi:10.1038/9195.
- ↑ Akopian AN, Sivilotti L & Wood JN (1996). "A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons". Nature. 379 (6562): 257–262. doi:10.1038/379257a0.
- ↑ Cummins TR, Sheets PL, Waxman SG (October 2007). "The roles of sodium channels in nociception: Implications for mechanisms of pain". Pain. 131 (3): 243–57. doi:10.1016/j.pain.2007.07.026. PMID 17766042.
- 1 2 Swanwick RS, Pristerá A & Okuse K (2010). "The trafficking of NaV1.8". Neuroscience Letters. 486: 78–83. doi:10.1016/j.neulet.2010.08.074.
- ↑ Strickland IT, Martindale JC, Woodhams PL, Reeve AJ, Chessell IP & McQueen DS (2008). "Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain". European Journal of Pain. 12: 564–572. doi:10.1016/j.ejpain.2007.09.001.
- ↑ Blair NT & Bean BP (2002). "Roles of Tetrodotoxin (TTX)-Sensitive Na+ Current, TTX-Resistant Na+ Current, and Ca2+ Current in the Action Potentials of Nociceptive Sensory Neurons". The Journal of Neuroscience. 22 (23): 10277–10290. PMID 12451128.
- ↑ Renganathan M, Cummins TR & Waxman SG (2001). "Contribution of Nav1.8 Sodium Channels to Action Potential Electrogenesis in DRG Neurons". Journal of Neurophysiology. 86 (2): 629–640. PMID 11495938.
- ↑ Millan MJ (1999). "The induction of pain: an integrative review". Progress in Neurobiology. 57: 1–164. doi:10.1016/S0301-0082(98)00048-3.
- ↑ Matthews EA, Wood JN & Dickenson AH (2006). "Nav 1.8-null mice show stimulus-dependent deficits in spinal neuronal activity". Molecular Pain. 2: 5. doi:10.1186/1744-8069-2-5.
- ↑ Jarvis MF, Honore P, Shieh C, et al. (2007). "A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat". PNAS. 104: 8520–8525. doi:10.1073/pnas.0611364104. PMC 1895982
 . PMID 17483457.
. PMID 17483457.
- 1 2 Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JW & Reeh PW (2007). "Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures". Nature. 447: 856–859. doi:10.1038/nature05880.
- ↑ Devor M; Govrin-Lippmann R & Angelides (1993). "Na+ Channel lmmunolocalization in Peripheral Mammalian Axons and Changes following Nerve Injury and Neuroma Formation". The Journal of Neuroscience. 13 (5): 1976–1992. PMID 7683047.
- ↑ Gold MS, Reichling DB, Shuster MJ & Levine JD (1996). "Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current innoclceptors". PNAS. 93 (3): 1108–1112. doi:10.1073/pnas.93.3.1108. PMC 40039. PMID 8577723.
- ↑ England S, Bevan S & Docherty RJ (1996). "PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade". The Journal of Physiology. 1: 492–440.
- ↑ Liu C, Su Y & Bao L (2010). "Prostaglandin E2 Promotes Nav1.8 Trafficking via Its Intracellular RRR Motif Through the Protein Kinase A Pathway". Traffic. 11: 1–666. doi:10.1111/j.1600-0854.2009.01027.x.
- ↑ Khasar SG, Gold MS & Levine JD (1998). "A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat". Neuroscience Letters. 30 (1): 17–20. PMID 9832206.
- ↑ Wu D, Chandra D, McMahon T, Wang D, Dadgar J, Kharazia VN, Liang Y, Waxman SG, Dib-Hajj SD & Messing RO (2012). "PKCε phosphorylation of the sodium channel NaV1.8 increases channel function and produces mechanical hyperalgesia in mice". J Clin Invest. 122: 1306–1315. doi:10.1172/JCI61934.
- ↑ Hu, D; Barajas-Martínez, H; Pfeiffer, R; Dezi, F; Pfeiffer, J; Buch, T; Betzenhauser, M. J.; Belardinelli, L; Kahlig, K. M.; Rajamani, S; Deantonio, H. J.; Myerburg, R. J.; Ito, H; Deshmukh, P; Marieb, M; Nam, G. B.; Bhatia, A; Hasdemir, C; Haïssaguerre, M; Veltmann, C; Schimpf, R; Borggrefe, M; Viskin, S; Antzelevitch, C (2014). "Mutations in SCN10A Are Responsible for a Large Fraction of Cases of Brugada Syndrome". Journal of the American College of Cardiology. 64 (1): 66–79. doi:10.1016/j.jacc.2014.04.032. PMID 24998131.
- ↑ McMahon SB (1984). "NGF as a Mediator of Inflammatory Pain". Phil. Trans. R. Soc. Lond. 351: 431–440. doi:10.1098/rstb.1996.0039.
- ↑ Okuse K, Malik-Hall M, Baker MD, Poon L, Kong H, Chao MV & Wood JN (2002). "Annexin II light chain regulates sensory neuron-specific sodium channel expression". Nature. 417 (6889): 653–656. doi:10.1038/nature00781. PMID 12050667.
- ↑ Foulkes T, Nassar MA, Lane T, Matthews EA, Baker MD, Okuse K, Dickenson AH & Wood JN (2006). "Deletion of Annexin 2 Light Chain p11 in Nociceptors Causes Deficits in Somatosensory Coding and Pain Behavior". The Journal of Neuroscience. 26: 10499–10507. doi:10.1523/JNEUROSCI.1997-06.2006.
- 1 2 Pristerà A, Baker MD & Okuse K (2012). "Association between Tetrodotoxin Resistant Channels and Lipid Rafts Regulates Sensory Neuron Excitability". PLoS One. 7: e40079. doi:10.1371/journal.pone.0040079.
- ↑ Hoeijmakers JG, Faber CG, Merkies IS & Waxman SG (2012). "Small-fibre neuropathies—advances in diagnosis, pathophysiology and management". Nature Reviews Neurology. 8: 369–379. doi:10.1038/nrneurol.2012.97.
- ↑ Faber CG, Hoeijmakers GK, Ahn H, Cheng X, Han C, Choi J, Lauria G, Vanhoutte EK, Gerrits MM, Dib-Hajj S, Drenth JPH, Waxman SG & Merkies ISJ (2012). "Gain of function NaV1.7 mutations in idiopathic small fiber neuropathy". Annals of Neurology. 71: 26–39. doi:10.1002/ana.22485.
Further reading
- Okuse K (2007). "Pain signalling pathways: From cytokines to ion channels". The International Journal of Biochemistry & Cell Biology. 39: 490–496. doi:10.1016/j.biocel.2006.11.016.
- Waxman SG (2013). "Painful Na-channelopathies: an expanding universe". Trends in Molecular Medicine. 19: 406–409. doi:10.1016/j.molmed.2013.04.003.
- Lai J, Porreca F, Hunter JC & Gold MS (2004). "Voltage-gated sodium channels and hyperalgesia". Annual Review of Pharmacology and Toxicology. 44: 371–397. doi:10.1146/annurev.pharmtox.44.101802.121627.
- Wood JN, Boorman JP, Okuse K, Baker MD (2004). "Voltage-gated sodium channels and pain pathways". Journal of Neurobiology. 61: 55–71. doi:10.1002/neu.20094.
- Malik-Hall M, Poon WY, Baker MD, et al. (2003). "Sensory neuron proteins interact with the intracellular domains of sodium channel NaV1.8.". Brain Res. Mol. Brain Res. 110 (2): 298–304. doi:10.1016/S0169-328X(02)00661-7. PMID 12591166.
- Yamaoka K, Inoue M, Miyazaki K, et al. (2009). "Synthetic Ciguatoxins Selectively Activate Nav1.8-derived Chimeric Sodium Channels Expressed in HEK293 Cells". J. Biol. Chem. 284 (12): 7597–605. doi:10.1074/jbc.M806481200. PMC 2658054. PMID 19164297.
- Choi JS, Hudmon A, Waxman SG, Dib-Hajj SD (2006). "Calmodulin regulates current density and frequency-dependent inhibition of sodium channel Nav1.8 in DRG neurons". J. Neurophysiol. 96 (1): 97–108. doi:10.1152/jn.00854.2005. PMID 16598065.
- Liu CJ, Priest BT, Bugianesi RM, et al. (2006). "A high-capacity membrane potential FRET-based assay for NaV1.8 channels". ASSAY and Drug Development Technologies. 4 (1): 37–48. doi:10.1089/adt.2006.4.37. PMID 16506887.
- Browne LE, Blaney FE, Yusaf SP, et al. (2009). "Structural Determinants of Drugs Acting on the Nav1.8 Channel". J. Biol. Chem. 284 (16): 10523–36. doi:10.1074/jbc.M807569200. PMC 2667739. PMID 19233853.
- Rabert DK, Koch BD, Ilnicka M, et al. (1998). "A tetrodotoxin-resistant voltage-gated sodium channel from human dorsal root ganglia, hPN3/SCN10A". Pain. 78 (2): 107–14. doi:10.1016/S0304-3959(98)00120-1. PMID 9839820.
- Plummer NW, Meisler MH (1999). "Evolution and diversity of mammalian sodium channel genes". Genomics. 57 (2): 323–31. doi:10.1006/geno.1998.5735. PMID 10198179.
- Catterall WA, Goldin AL, Waxman SG (2005). "International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels". Pharmacol. Rev. 57 (4): 397–409. doi:10.1124/pr.57.4.4. PMID 16382098.